Cell Ranger7.1, printed on 03/27/2025
Adaptive immunity is based on clonal selection and expansion from a repertoire of T and B lymphocytes bearing a diversity of cell-surface receptors and antibodies that recognize specific antigens. The enormous diversity of lymphocyte receptors and antibodies expressed from a relatively small number of gene segments is largely made possible through V(D)J recombination. The hypervariable recombined V(D)J transcripts in turn determine the affinity of the Complementary Determining Regions (CDRs) to bind to specific epitopes. See the Glossary for more details.
The 10x Genomics 5' Chromium Next GEM Single Cell Immune Profiling Solution enables simultaneous analysis of these libraries at single cell resolution for the same set of cells:
This makes the Immune Profiling Solution ideal for understanding the key features of an adaptive immune response and their impact on other genomic analytes, disease severity, therapy, and outcome.
Paired T or B cell receptor sequences are obtained per cell by partitioning thousands of cells into nanoliter-scale Gel Beads-in-Emulsion (GEMs). All cDNA generated in a single GEM share a common 10x Barcode. Inside a GEM, one to several UMIs are captured for each V(D)J chain. A round of enrichment PCR targeting the 5′ end to the C-region, followed by enzymatic fragmentation results in a pool of molecules originating from the same transcript. The molecules carry the same 10x Barcode and UMI sequences, but with different insert lengths, resulting in different sequence start points. The diversity of start points gives complete coverage of the targeted portion of each V(D)J transcript, which is typically ~650 bp.
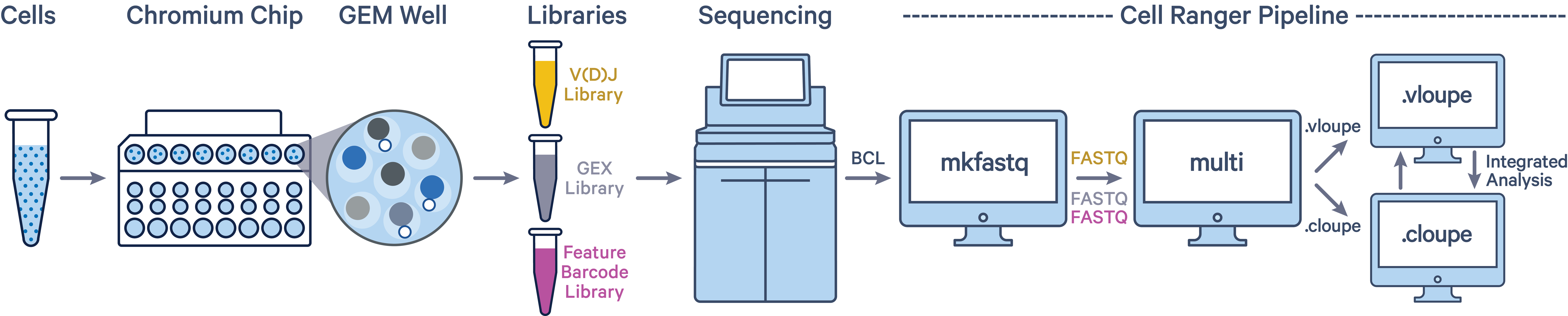
Analysis of these data types can be performed using the latest versions of Cell Ranger. The following illustration depicts how the cellranger multi pipeline can be used to analyze FASTQ data derived from these three Immune Profiling library types from the same GEM Well:
Skip Cell Ranger download and installation and get started with 10x Genomics Cloud Analysis, our recommended method for running Cell Ranger pipelines for most new customers. Use your web browser to easily generate Cell Ranger outputs from your FASTQ files and aggregate outputs from multiple runs, free for every 10x Genomics sample. Currently available only in the United States and Canada. Sign up for a free account or view tutorials and learn more.
Learn how to install and run Cell Ranger.
Cell Ranger includes five pipelines relevant to Immune Profiling data analysis:
cellranger mkfastq demultiplexes raw base call (BCL) files generated by Illumina sequencers into FASTQ files. It is a wrapper around Illumina's bcl2fastq, with additional useful features that are specific to 10x Genomics libraries and a simplified sample sheet format.
cellranger vdj takes FASTQ files from cellranger mkfastq or bcl2fastq for V(D)J libraries and performs sequence assembly and paired clonotype calling. It uses the Chromium cellular barcodes and UMIs to assemble V(D)J transcripts per cell. Clonotypes and CDR3 sequences are output as a .vloupe file which can be loaded into Loupe V(D)J Browser.
cellranger count takes FASTQ files from cellranger mkfastq or bcl2fastq for 5' Gene Expression and/or Feature Barcode (Antibody Capture, and/or CRISPR Guide Capture) libraries and performs alignment, filtering, barcode counting, and UMI counting. It uses the Chromium cellular barcodes to generate feature-barcode matrices, determine clusters, and perform gene expression analysis. The cellranger count pipeline outputs a .cloupe file which can be loaded into Loupe Browser for interactive visualization, clustering, and differential expression analysis.
cellranger multi takes FASTQ files from cellranger mkfastq or bcl2fastq for any combination of 5' Gene Expression, Feature Barcode (Antibody Capture, CRISPR Guide Capture, and/or Antigen Capture), and V(D)J libraries from a single GEM well. It performs alignment, filtering, barcode counting, and UMI counting on the Gene Expression and/or Feature Barcode libraries. It also performs sequence assembly and paired clonotype calling on the V(D)J libraries. Additionally, the cell calls provided by the Gene Expression data are used to improve the cell calls inferred by the V(D)J library.
cellranger aggr aggregates outputs from multiple runs of cellranger count cellranger vdj, or cellranger multi. The aggr pipeline normalizes the individual Gene Expression and Feature Barcode runs to the same sequencing depth, recomputes the feature-barcode matrices and performs analysis on the combined data. The pipeline also recomputes the V(D)J clonotype grouping on the combined data.
cellranger reanalyze takes feature-barcode matrices produced by cellranger count, cellranger multi, cellranger vdj, or cellranger aggr and reruns the dimensionality reduction, clustering, and gene expression algorithms using tunable parameter settings.
The .cloupe files and the .vloupe files output by the pipelines can be overlaid for an integrated analysis with Loupe Browser and Loupe V(D)J Browser. Refer to Analysis of Multiple Library Types for more information.
The libraries supported by different versions of Cell Ranger:
| Single Cell Immune Profiling Solution | CR 7.1 | CR 7.0 | CR 6.1 | CR 6.0 | CR 5.0 | CR 4.0 | CR 3.1 | CR 3.0 | CR 2.2 |
|---|---|---|---|---|---|---|---|---|---|
| 5’ Gene Expression | |||||||||
| V(D) J | |||||||||
| 5’ Gene Expression + V(D) J | |||||||||
| 5’ Gene Expression + Cell Surface Protein | |||||||||
| 5’ Gene Expression + Cell Surface Protein + V(D) J | |||||||||
| V(D) J Enriched + Cell Surface Protein | |||||||||
| 5’ Cell Surface Protein only | |||||||||
| Targeted Gene Expression | |||||||||
| 5’ HT (High Throughput) | |||||||||
| 5’ Gene Expression + CRISPR | |||||||||
| 5’ Gene Expression + CRISPR + Cell Surface Protein | |||||||||
| 5’ Gene Expression + CRISPR + V(D)J Enriched Libraries | |||||||||
| 5’ Gene Expression + Antigen Capture + V(D)J ± Cell Surface Protein |