Cell Ranger7.1, printed on 03/27/2025
The assembly process takes the reads for a single barcode as input. These reads are then glued together, outputting a set of assembled contigs that represent the best estimate of transcript sequences present. Each base in each contig is assigned a quality value. The numbers of UMIs and reads supporting each contig are also tracked.
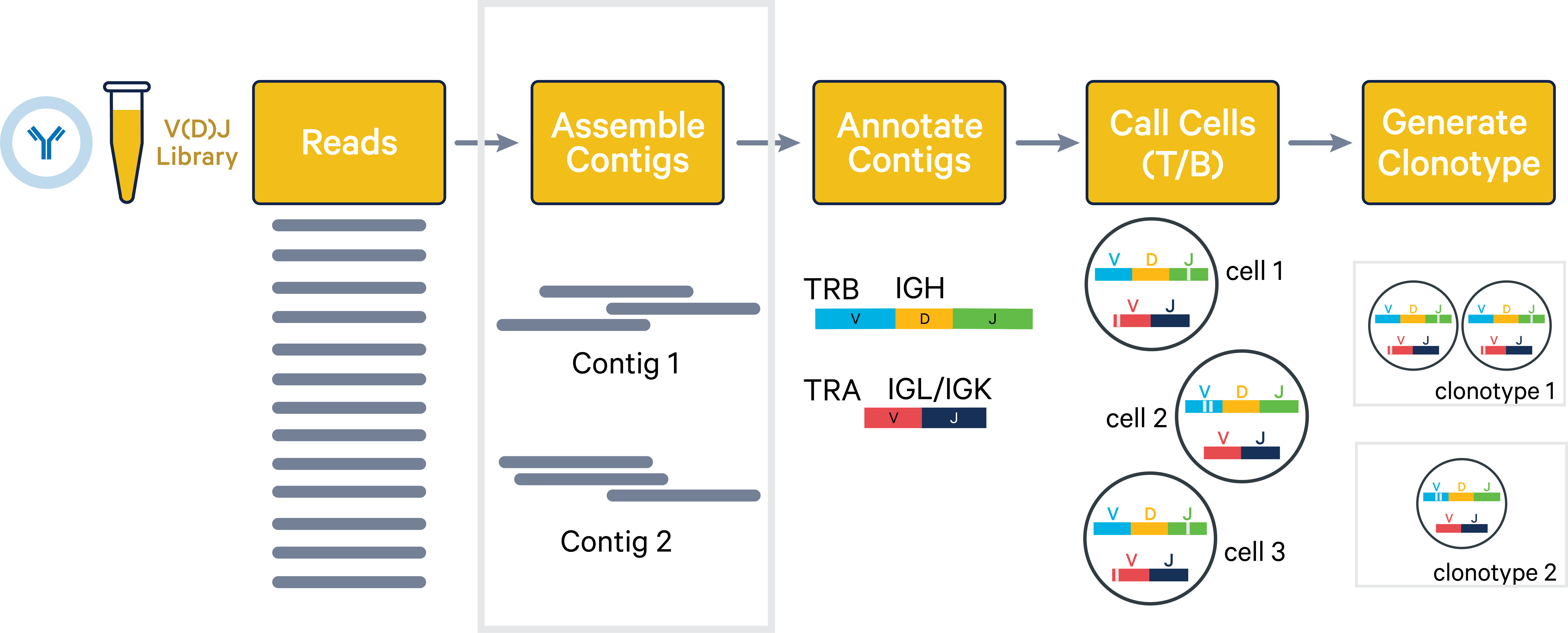
The assembler uses the V(D)J reference sequence during assembly, unless the pipeline is run in de novo mode. Parts of the Annotation Algorithm page may be relevant to learn more about more about the assembly process.
Contig assembly is complicated by noise that can arise from many sources. Some sources of noice include:
| Step | Operation |
|---|---|
| Adapter trimming | Trim adapters using a custom algorithm. |
| Read subsampling | Downsample reads for a given barcode to retain a maximum of 80,000 reads. >80,000 reads do not improve results. |
| Read trimming | Trim off nucleotides in the read after the enrichment primers. |
| Graph formation | Build a De Bruijn graph using kmer length (k) = 20 |
| Reference-free graph simplification | Simplify the graph by removing noisy edges. |
| Reference-assisted graph simplification | Use the V(D)J reference to remove noisy edges. |
| UMI filtering | Filter out UMIs that are likely to be artifacts. |
| Contig construction | Build contigs by looking for the best path through the graph for each UMI. |
| Competitive deletion of contigs | Compare contigs, remove weak contigs that are likely to be artifacts. |
| Contig confidence | Define high confidence contigs that are likely to represent bona fide transcripts from a single cell (associated to one barcode). |
| Contig quality scores | Assign a quality score to each base on each contig. |
Known adapter and primer sequences from the 5’ and 3’ ends of reads are trimmed using a custom 10x Genomics trimming tool.
Some cells have extremely high coverage. High coverage could be either due to true high sequencing coverage, or high mRNA expression in plasma cells (commonly seen in BCR).
Very high coverage (greater than 80,000 reads) of transcripts can be problematic because it degrades computational performance and adds little information. Therefore, coverage is capped to a maximum of 80,000 reads per barcode. If there are more than 80,000 reads for a any given 10x Barcode, the reads are downsampled.
The inner enrichment primers hybridize to constant regions of V(D)J genes. Any bases to the right of those positions should not be present in the data. They are trimmed from the reads.
A De Bruijn graph using k = 20 is created and transformed into a directed graph. The edges of the graph are DNA sequences corresponding to unbranched paths in the De Bruijn graph.
A collection of heuristic steps is applied to simplify the graph. During this process read support on each edge is tracked and edited. Several examples of simplification steps are described:
For each branch in the graph, and for each UMI, if one branch has ten times more reads than a second branch, read support for the UMI from the second branch is removed.
When two branches emanate from a vertex, the weaker branch is deleted based on these criteria:
(a) There are at least twice as many reads on the strong branch.
(b) There are fewer than 8 reads for any UMI on the weak branch.
(c) For every UMI, the strong branch has twice as many reads as the weak branch with utmost one exception (such as events like alternate splicing) where the event is supported by only one UMI.
Path cleaning: For each UMI, the strongest path is defined. Then graph edges that are not on this path are deleted.
Component cleaning: For each UMI, if one graph component has ten times more reads supporting it than a second component, the read support for the second component is deleted.
If the pipeline is run in reference-assisted mode (not de novo assembly), bubbles in the graph are popped with the aid of the reference sequence. There are several heuristic tests, all of which require that both bubble branches have the same length. An example scenario is when branch 1 is supported by at least three UMIs and has a kmer matching the reference, whereas branch 2 is supported by a single UMI, and has no kmers matching the reference. In this scenario, the weaker branch (branch 2) is deleted.
UMIs that survive these filtration steps are retained:
Find the single strongest path for each UMI. A strong path either contains a reference kmer, or if assembled de novo, matches a primer (described above).
Find good graph edges that appear on one or more strong paths.
Sort the reads based on these good graph edge assignments.
Find the UMIs for these reads.
Remove any UMI for which less than 50% of kmers are contained in good edges. are contained in good edges.
For reference-assisted assembly, if none of the strong paths had a V segment annotation, remove all the UMIs for that barcode.
Initially, every strong path that either contains an enrichment primer (de novo assembly) or is annotated by a CDR3 (in the reference-assisted assembly) is called a contig.
Then, in reference-assisted assembly:
Contigs are trimmed to remove nucleotides occurring before the 5' UTR for a V segment and after enrichment primers.
Contigs that have only a C annotation are deleted. These deleted contigs are enriched for artifacts.
If a contig has a single-base indel relative to the reference that is supported by a single UMI (or one UMI plus one additional read), the indel is corrected to reflect the reference sequence.
Contigs with fewer than 300 base pairs are removed.
At this stage in assembly, there can be some redundancy among contigs arising from actual differences in transcripts, laboratory technical artifacts, or artifacts in contig construction.
Steps to eliminate redundancy:
The number of UMIs assigned to each contig is computed.
Junction selection:
Competitive deletion of contigs aims to delete contigs that arise from extracellular mRNA in the sample or other background processes.
For reference-assisted assembly, the junction sequence of each productive contig is defined to be 100 nucleotides at the end of the annotated J segment. The junction UMI support for the contig is the number of UMIs that cover the junction sequence. Reads that support the junction sequence make up the junction read support. Suppose we have two contigs with respective (junction UMI support, junction read support) = (u1,n1) and (u2,n2). Suppose that (u1,n1) is sufficiently larger than (u2,n2). For example, u1 ≥ 2, u2 = 1, n1 ≥ 2 * n2 would qualify. (And there are some similar criteria, not listed here.) Then if the contigs have the same chain type, we delete the second contig.
In de novo mode, a similar criterion is applied to contigs containing a CDR3, but instead of the junction mode used in the reference-assisted assembly, the 100 nucleotides starting at the end of the CDR3 are used. Chain type is not considered when deleting a contig, and the two strongest contigs are protected from deletion.
Incorrect clonotypes can arise from sources such as extracellular mRNA or doublets. To prevent this, the confidence of a contig is assessed and those declared low confidence are excluded. All non-productive contigs are declared to have low confidence. For reference-assisted assembly, productive contigs have low confidence if any of the following apply:
There are more than four productive contigs.
There are more than two contigs for a chain type (e.g. three TRA contigs): It is expected that each cell-associated barcode has one productive TRA and one productive TRB chain for T cells, and one productive heavy and one productive light chain for B cells. Extra productive contigs are less likely to be legitimate.
There are less than three filtered UMIs each having at least three read pairs
All productive contigs have junction UMI support at most one, and either there are less than five filtered UMIs each having at least three read pairs, or there are more than two productive contigs
Some productive contig has junction UMI support at most one, and there are less than three filtered UMIs each having at least n/20 read pairs, where n is the N50, taken over all UMIs in the entire dataset that have at least two read pairs, of the number of read pairs in such UMIs
Individual productive contigs are downgraded if their junction UMI support is at most one and the number of productive contigs exceeds two.
In de novo mode, similar criteria are applied. Here, 'productive contig' is replaced by 'contig having a CDR3 sequence'. The chain type test is not applied.
Each base in the assembled contig is assigned a Phred-scaled quality value (QV), representing an estimate of the probability of an error at that base. The QV is computed with a hierarchical model that accounts for the errors in:
The sequencing error model uses the reported sequencer QVs. At recommended sequencing depths, many reads per UMI are observed. This allows for sequencing errors in individual reads to be corrected rapidly.
The estimated error rate for the V(D)J RT reaction is 1e-4 per base. Therefore, assembled bases that are covered by a single UMI are assigned Q40, and bases covered by at least two UMIs are assigned Q60.