Cell Ranger7.1, printed on 04/22/2025
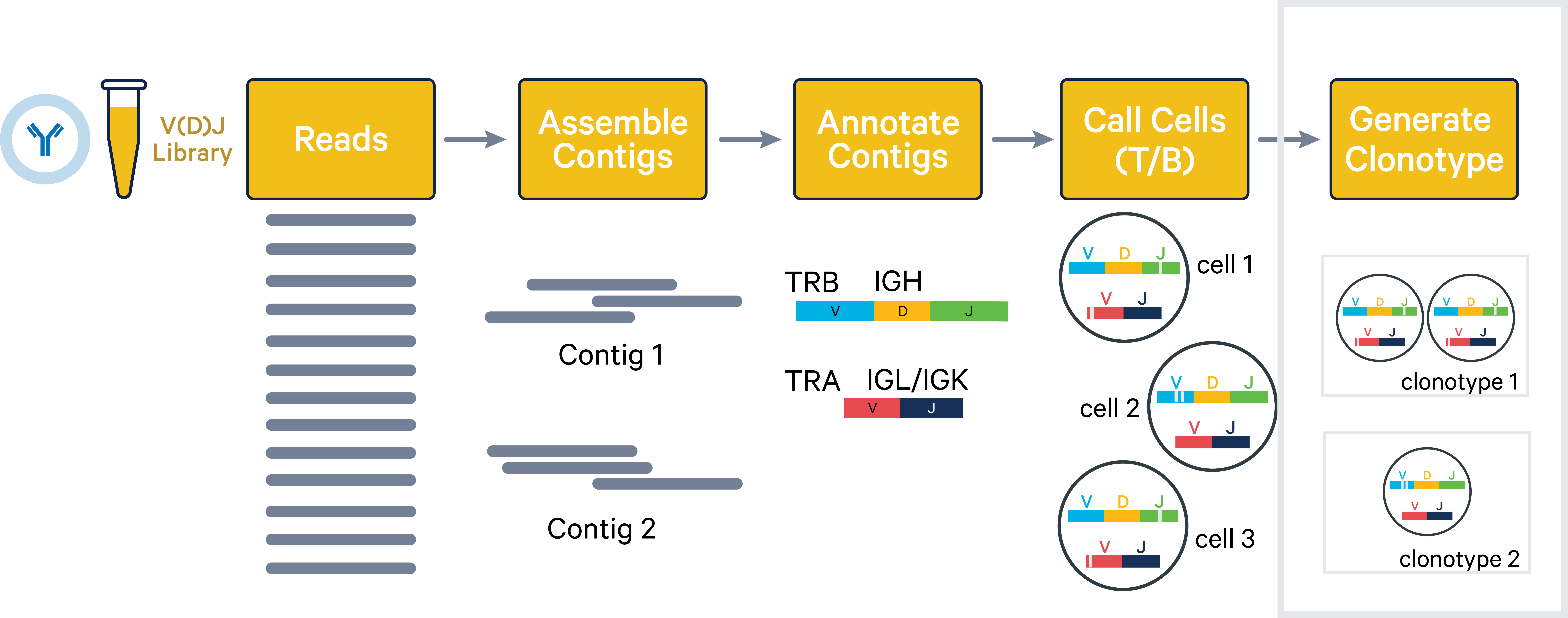
During the clonotype grouping stage, cell barcodes are placed in groups called clonotypes. Each clonotype consists of all descendants of a single, fully rearranged common ancestor, as approximated computationally. During this process, some cell barcodes are flagged as likely artifacts and filtered out, meaning that they are no longer called as cells.
T cells: The lack of somatic hypermutation (SHM) in T cell receptors (TCRs) yields biological clonotypes that have identical V(D)J transcripts. Technical artifacts (e.g. arising in reverse transcription) can result in the computed clonotypes having isolated differences. These are rare.
B cells: Fully rearranged B cell receptors (BCRs) can undergo SHM, which can increase antigen affinity. Thus for BCRs, V(D)J transcripts in a clonotype can differ at any position, as shown below:
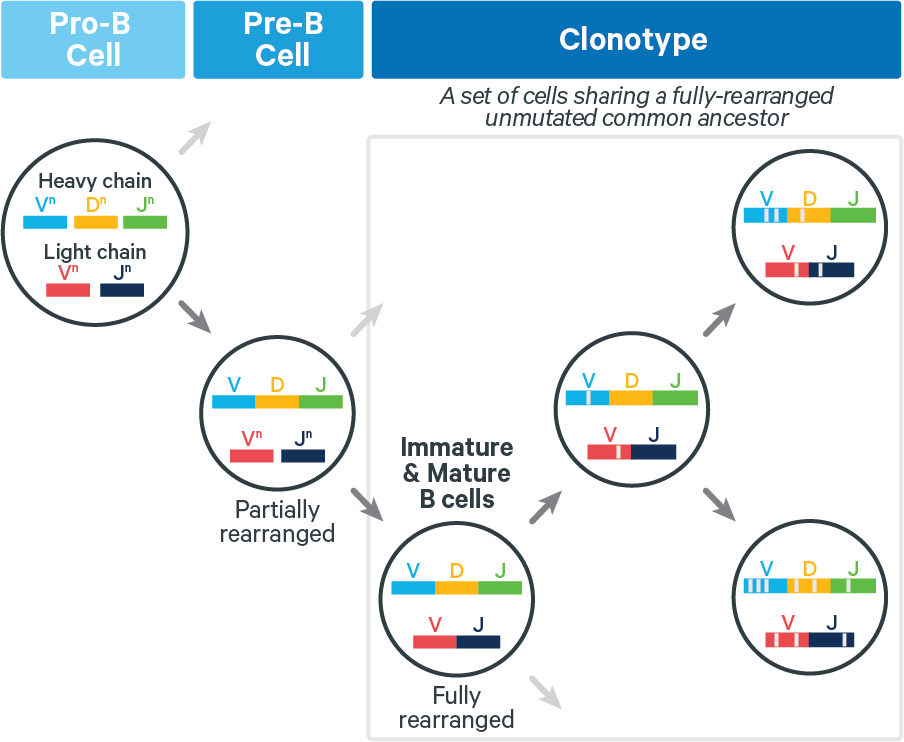
B cell clonotypes can be hard to infer accurately because SHM can introduce numerous mutations. Cell Ranger v5.0 and above accomplishes B cell clonotype grouping by invoking a module for clonal analysis called enclone which simultaneously filters and groups cells into clonotypes.
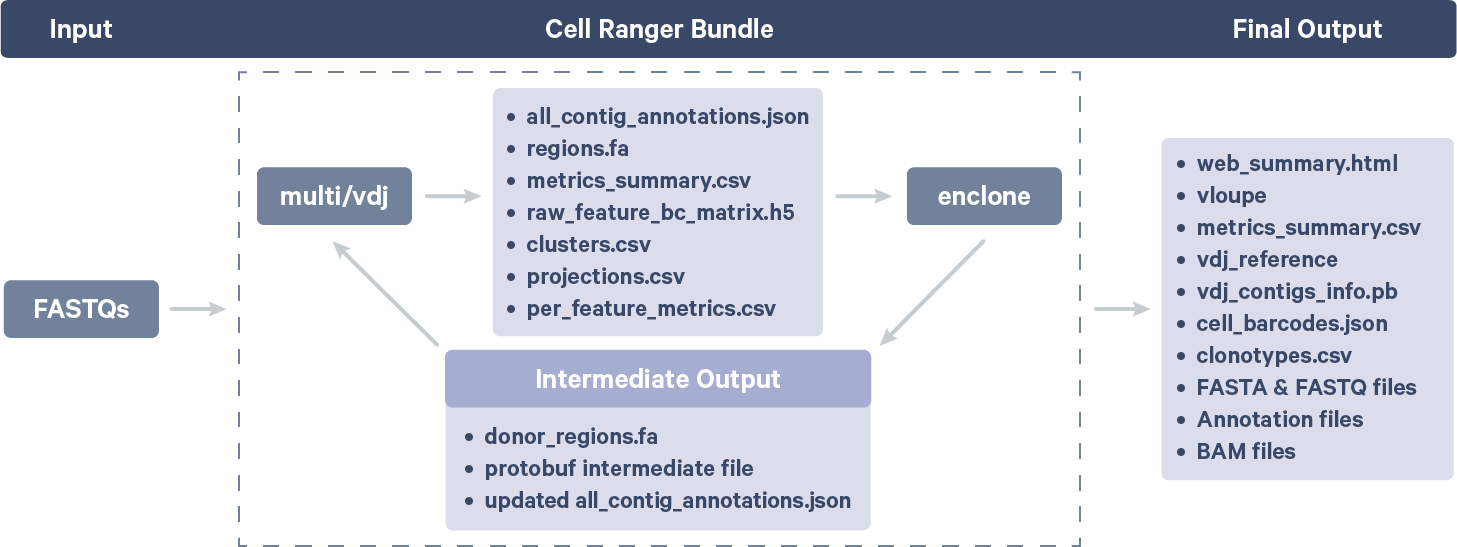
For clonotype grouping, enclone mainly gets its information from an internally generated Cell Ranger file called all_contig_annotations.json. The relationship between enclone and Cell Ranger is illustrated here:
For each dataset, enclone derives the reference sequence for V genes in the donor's genome (germline sequence) to use as a reference for SHMs.
Germline variant assessment for J genes is currently not performed as it does not greatly enhance clonotype specificity.
Cells are placed into groupings called exact subclonotypes if they have identical V(D)J transcripts. Only productive contigs are used. Exact subclonotypes must have the same number of chains. They must also be identical in their V(D)J sequences and constant region gene assignments. Exact subclonotypes are not required to have identical 5' UTRs. Additionally, the algorithm does not test for SHM in the 5' UTR or constant region.
Exact subclonotypes are iteratively merged into clonotypes based on comparing each pair of exact subclonotypes to each other. Two cells with set criteria of shared differences and minimal CDR3 mutations are deemed to be in the same clonotype. Merging criteria are briefly described here. Visit the enclone help page for details.
During library generation, artifacts can arise by two mechanisms:
(a) Reverse transcription or sequencing can introduce base call errors. These usually occur at bases having low quality scores. Cells with these low quality bases are screened out, typically at a low rate.
(b) Gel Beads-in-emulsion (GEMs) may contain material from two or more cells: entire intact cells, cell fragments, or individual mRNA molecules.
Contamination detection is a complex task and is accomplished via multiple heuristic filters. Some barcode filtering happens during the assembly and cell calling stages of Cell Ranger execution. enclone uses Cell Ranger's cell calling information and additionally performs its own barcode and clonotype filtering by employing a series of heuristic tests. Filtering and clonotype grouping happen simultaneously.
Within Cell Ranger, enclone applies these default filters, some of which are recursive:
| Filter | Description |
|---|---|
| Cell filter | Remove barcodes not called cells in cellranger vdj pipeline. |
| Maximum contigs filter | Remove barcodes with more than four productive contigs. |
| Graph filter | Remove some exact subclonotypes that appear to be background. |
| Cross filter | Use cross-library information (i.e., from two libraries originating from the same donor) To remove spurious exact subclonotypes. |
| Barcode duplication filter | Remove duplicated barcodes within an exact subclonotype. |
| Whitelist filter | Identify and remove any artifactual barcodes that do not match a barcode in the 10x Genomics barcode whitelist. Artifactual barcodes are rare and likely arise from Gel Bead contamination. |
| Foursie filter | Remove some four-chain clonotypes that are biologically irrelevant, e.g., 4 heavy chains. |
| Improper filter | Remove exact subclonotypes having 3 or 4 identical chains. |
| Weak onesie filter | Disintegrate some single-chain clonotypes into single cells. If a barcode has a high confidence contig, passes the cell calling filter, and has only 1 chain, it is retained as its own clonotype. |
| UMI filter | Determined a baseline UMI count for each dataset and remove any B cells having UMI counts lower than this baseline. Helps eliminate rare clonotype expansion signatures arising from fragmentation of plasma cells or other poorly understood physical processes. |
| UMI ratio filter | Remove some B cells with low UMI counts, relative to mean UMI counts in a given clonotype. |
| GEX filter | If using cellranger multi, remove barcodes that were called as cells in the V(D)J but not the GEX library. This filter mitigates any overcalling issues seen in BCR and TCR libraries. |
| Doublet filter | Remove some barcodes that appear to represent doublets or higher-order multiplets |
| Signature filter | Some complex clonotypes with many chains represent multiple true clonotypes that are glued together into a single clonotype. This filter removes some exact subclonotypes that appear to represent contaminants, based on their chain signature. |
| Onesie merger | Prevent the merger of some single-chain clonotypes into other clonotypes. |
| Weak chain filter | From the remaining cells, remove any cells that have weak chains. A chain is weak if it is found in ≤ 5 other cells, and the total number of cells in that clonotype is less than 5 times that number. E.g., if there are a total of 14 cells in a clonotype, and a given chain is found in only 3 of those cells, all 3 cells are filtered out. However, if there were at least 3 x 5 (15 cells) in the clonotype, the 3 cells with this chain would be retained. |
| Quality merger | Filter out exact subclonotypes with low quality score positions. |
Visit the enclone website for detailed documentation.
enclone is also available in beta for exploratory evaluation via the use of command line arguments, permitting granular control over clonotyping and filtering heuristics. enclone can also display clonotypes and infer phylogenetic trees.
enclone is open source, unsupported, and separate from Cell Ranger.