Cell Ranger7.1, printed on 03/26/2025
In this tutorial, you will:
To follow along, you must:
The Chromium Single Cell 5’ Barcode Enabled Antigen Mapping (BEAM) technology offers a scalable approach for mapping a V(D)J receptor to a target antigen by enabling the detection of gene expression profiles, paired V(D)J receptors, and signal from a bound antigen from the same single cell. All of these libraries, generated from a single GEM well, can be analyzed together with Cell Ranger v7.1 or later using the cellranger multi pipeline.
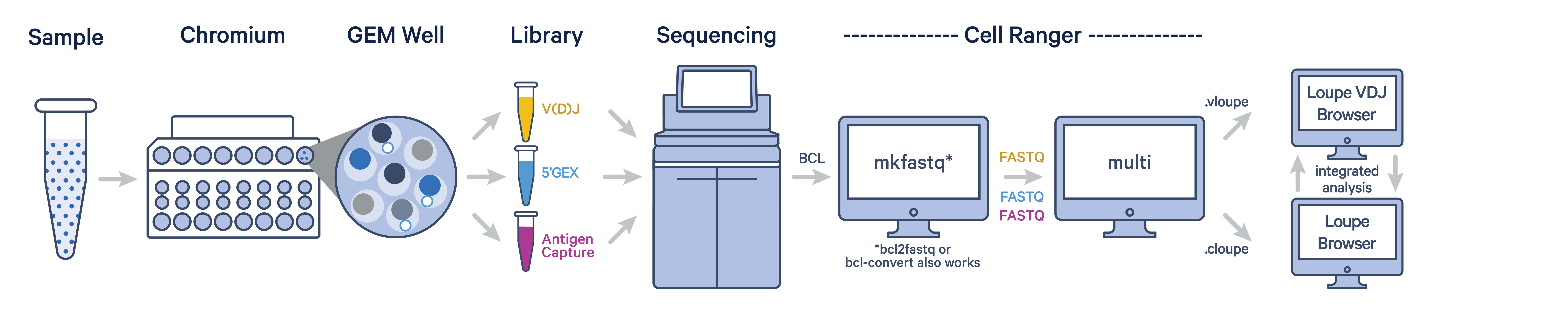
We will work with the 5k Human A0201 | B0702 PBMCs (BEAM-T) dataset.
Open up a terminal window. You may log in to a remote server or choose to perform the compute on your local machine. Refer to the System Requirements page for details.
In the working directory, create a new folder called beam-t and cd into that folder:
mkdir beam-t cd beam-t
Download the input FASTQ files:
curl -O https://cf.10xgenomics.com/samples/cell-vdj/7.1.0/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_fastqs.tar
A file named 5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_fastqs.tar should appear in your directory when you list files with the ls -lt command.
Uncompress the FASTQs:
tar -xf 5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_fastqs.tar
You should now see a folder called 5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_fastq
cd 5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_fastq ls
The folder contains three subfolders with library-specific FASTQS files: antigen_capture, gex, and vdj.
Navigate back to the working directory:
cd ..
Double check you are in the correct directory by running the ls command; the working directory should have the FASTQs 5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_fastqs folder.
Download the Feature Reference CSV available for this example dataset.
curl -O https://cf.10xgenomics.com/samples/cell-vdj/7.1.0/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_count_feature_reference.csv
To view the contents of the Feature Reference CSV, open it in your text editor of choice (e.g., nano)
nano 5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_count_feature_reference.csv
The contents should look like this:
id,name,read,pattern,sequence,feature_type,mhc_allele Flu_A0201,Flu_A0201,R2,^(BC),GATTGGCTACTCAAT,Antigen Capture,HLA-A*02:01 CMV_B0702,CMV_B0702,R2,^(BC),CGGCTCACCGCGTCT,Antigen Capture,HLA-B*07:02 negative_control_A0201,negative_control_A0201,R2,^(BC),CTATCTACCGGCTCG,Antigen Capture,HLA-A*02:01 negative_control_B0702,negative_control_B0702,R2,^(BC),CATGTCTACGTTAAG,Antigen Capture,HLA-B*07:02
Since this is a BEAM-T (TCR Antigen Capture) dataset, the Feature Reference CSV contains the additional mhc_allele column. The BEAM-Ab tutorial guides you through analyzing a BCR Antigen Capture dataset.
When working with your own dataset, you must customize this file for your experiment. Learn more about the Feature Reference CSV.
Download the pre-built human reference transcriptome to the working directory (beam-t/) and uncompress it:
curl -O https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz tar -xf refdata-gex-GRCh38-2020-A.tar.gz
Next, download the pre-built V(D)J reference to the working directory and uncompress it:
curl -O https://cf.10xgenomics.com/supp/cell-vdj/refdata-cellranger-vdj-GRCh38-alts-ensembl-7.1.0.tar.gz tar -xf refdata-cellranger-vdj-GRCh38-alts-ensembl-7.1.0.tar.gz
In your working directory, create a new CSV file called 5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_config.csv using your text editor of choice. For example, you can create a file with nano using this command:
nano 5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_config.csv
Copy and paste this text into the newly created file and customize the /path/to/... part of file paths:
[gene-expression] ref,/path/to/references/refdata-gex-GRCh38-2020-A [feature] ref,/path/to/feature_references/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_count_feature_reference.csv [vdj] ref,/path/to/references/vdj/refdata-cellranger-vdj-GRCh38-alts-ensembl-7.1.0 [libraries] fastq_id,fastqs,lanes,feature_types beamt_human_A0201_B0702_pbmc_ag,/path/to/fastqs/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_fastqs/antigen_capture,1|2,Antigen Capture beamt_human_A0201_B0702_pbmc_vdj,/path/to/fastqs/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_fastqs/vdj,1|2,VDJ-T beamt_human_A0201_B0702_pbmc_gex,/path/to/fastqs/5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_fastqs/gex,1|2,Gene Expression [antigen-specificity] control_id,mhc_allele negative_control_A0201,HLA-A*02:01 negative_control_B0702,HLA-B*07:02
Use your text editor's save command to save the file. In nano, save by typing → → .
A customizable multi config CSV template is available for download on the example dataset page, under the Input Files tab.
Once you have all the necessary files, make a new directory called runs/ in your beam-t working directory:
mkdir runs/ cd runs/
You will run cellranger multi in the runs/ directory.
After downloading/creating the FASTQ files, Feature Reference CSV, reference transcriptome, and V(D)J reference, you are ready to run cellranger multi.
Print the usage statement to get a list of all the options:
cellranger multi --help
The output should look similar to:
user_prompt$ cellranger multi --help
cellranger-multi
Analyze multiplexed data or combined gene expression/immune profiling/feature
barcode data
USAGE:
cellranger multi [FLAGS] [OPTIONS] --id --csv
FLAGS:
--dry Do not execute the pipeline. Generate a pipeline
invocation (.mro) file and stop
--disable-ui Do not serve the web UI
--noexit Keep web UI running after pipestance completes or fails
--nopreflight Skip preflight checks
-h, --help Prints help information
OPTIONS:
--id A unique run id and output folder name [a-zA-Z0-
9_-]+
--description Sample description to embed in output files
[default: ]
--csv Path of CSV file enumerating input libraries and
analysis parameters
--jobmode Job manager to use. Valid options: local
(default), sge, lsf, slurm or path to a
.template file. Search for help on "Cluster
Mode" at support.10xgenomics.com for more
details on configuring the pipeline to use a
compute cluster [default: local]
--localcores Set max cores the pipeline may request at one
time. Only applies to local jobs
....
| Option | Description |
|---|---|
--id |
The id argument must be a unique run ID. We will call this run HumanB_Cell_multi based on the sample type in the example dataset. |
--csv |
Path to the multi config CSV file enumerating input libraries and analysis parameters. Your multi_config.csv file is in the working directory. When executing cellranger multi from the runs directory, the relative path should be: ../multi_config.csv |
From within the beam-t/runs/ directory, run cellranger multi
/path/to/cellranger-7.1.0/cellranger multi --id=beam-t-run --csv=../5k_BEAM-T_Human_A0201_B0702_PBMC_5pv2_Multiplex_config.csv
The run begins similarly to this:
user_prompt$ cellranger multi --id=beam-t-run --csv=/jane.doe/beam-t/multi_config.csv Martian Runtime - v4.0.10 2023-06-15 11:44:24 [jobmngr] WARNING: configured to use 334GB of local memory, but only 194.9GB is currently available. Serving UI at http://bespin3.fuzzplex.com:34513?auth=-Sm5gsg6_G8FjcUX0_YD5J8SYoBODz4IWoVIK9ec0jg Running preflight checks (please wait)... 2023-06-15 11:44:33 [runtime] (ready) ID.beam-t-run.SC_MULTI_CS.PARSE_MULTI_CONFIG 2023-06-15 11:44:33 [runtime] (run:local) ID.beam-t-run.SC_MULTI_CS.PARSE_MULTI_CONFIG.fork0.chnk0.main 2023-06-15 11:44:56 [runtime] (chunks_complete) ID.beam-t-run.SC_MULTI_CS.PARSE_MULTI_CONFIG 2023-06-15 11:44:56 [runtime] (ready) ID.beam-t-run.SC_MULTI_CS.FULL_COUNT_INPUTS.WRITE_GENE_INDEX 2023-06-15 11:44:56 [runtime] (run:local) ID.beam-t-run.SC_MULTI_CS.FULL_COUNT_INPUTS.WRITE_GENE_INDEX.fork0.chnk0.main ....
When the output of the cellranger multi command says, “Pipestance completed successfully!”, the job is done:
web_summary: /jane.doe/beam-t/runs/beam-t-run/outs/per_sample_outs/beam-t/web_summary.html
metrics_summary: /jane.doe/beam-t/runs/beam-t-run/runs/beam-t/outs/per_sample_outs/beam-t/metrics_summary$
}
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
A successful cellranger multi run produces a new directory called beam-t-run (based on the --id flag specified during the run). The contents of the beam-t-run directory:
. ├── beam-t-run │ ├── beam-t.mri.tgz │ ├── _cmdline │ ├── _filelist │ ├── _finalstate │ ├── _invocation │ ├── _jobmode │ ├── _log │ ├── _mrosource │ ├── outs │ ├── _perf │ ├── _perf._truncated_ │ ├── SC_MULTI_CS │ ├── _sitecheck │ ├── _tags │ ├── _timestamp │ ├── _uuid │ ├── _vdrkill │ └── _versions
The outs/ directory contains all important output files generated by the cellranger multi pipeline:
── runs
└── beam-t-run
└──outs
├── config.csv
├── multi
│ ├── count
│ │ ├── feature_reference.csv
│ │ ├── raw_cloupe.cloupe
│ ├── raw_feature_bc_matrix
│ │ ├── raw_feature_bc_matrix.h5
│ │ ├── raw_molecule_info.h5
│ │ ├── unassigned_alignments.bam
│ │ └── unassigned_alignments.bam.bai
│ └── vdj_t
│ ├── all_contig_annotations.bed
│ ├── all_contig_annotations.csv
│ ├── all_contig_annotations.json
│ ├── all_contig.bam
│ ├── all_contig.bam.bai
│ ├── all_contig.fasta
│ ├── all_contig.fasta.fai
│ └── all_contig.fastq
├── per_sample_outs
│ └── beam-t
│ ├── antigen_analysis
│ ├── count
│ ├── metrics_summary.csv
│ ├── vdj_t
│ └── web_summary.html
└── vdj_reference
├── fasta
│ ├── donor_regions.fa
│ └── regions.fa
└── reference.json