Cell Ranger7.1, printed on 04/25/2025
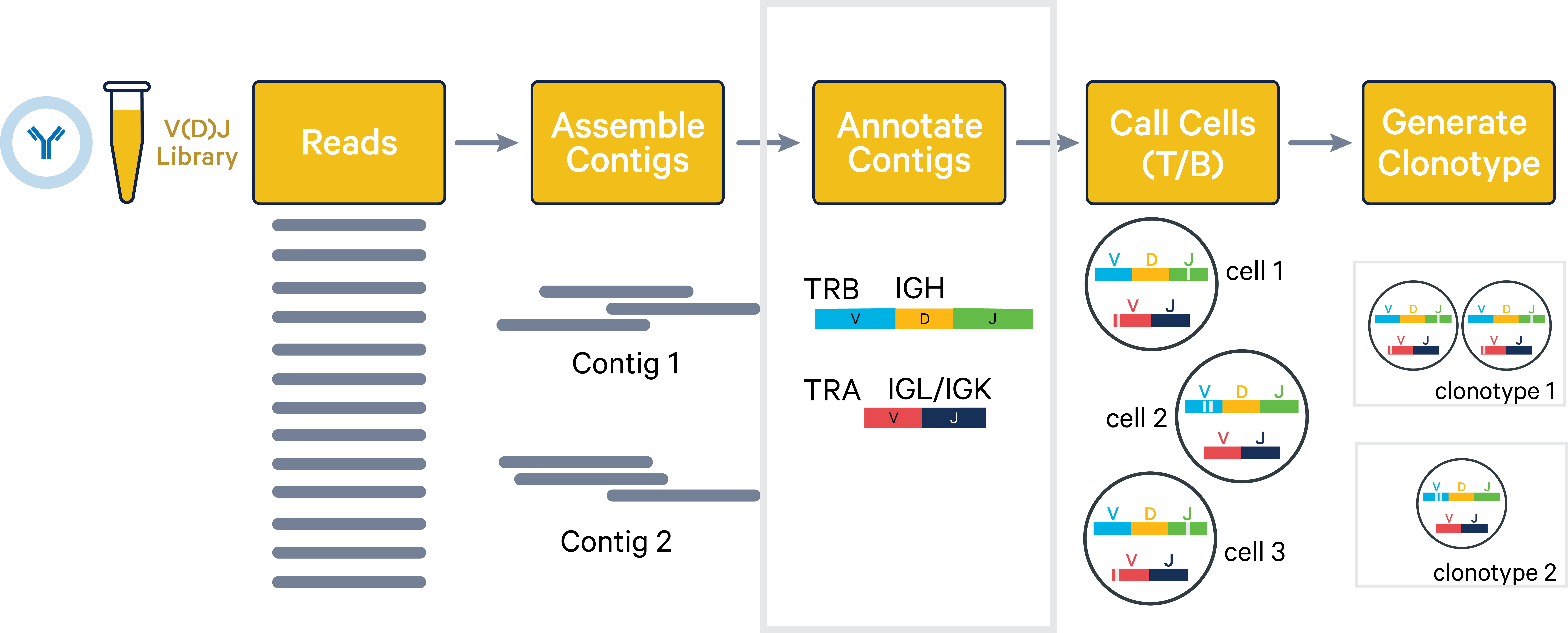
The three goals of V(D)J contig annotation are to 1) define the alignments of V, D, and J segments to a contig, 2) identify CDR3 sequences, and 3) from these data determine if a contig is productive, meaning that it is likely to correspond to a functional T or B cell receptor.
Cell Ranger first determines if the data are TCR or BCR. Then it aligns all contigs to the corresponding (TCR or BCR) reference sequences. Occasionally, contigs are aligned to both references. Alignment is seeded on 12-mer perfect matches, followed by heuristic extension. Cell Ranger also searches backwards from C segment alignments for J segment alignments that do not have 12-mer perfect matches, as these will arise occasionally from somatic hypermutation.
The choice of V(D)J reference sequences in an alignment can be arbitrary, depending on how similar the reference sequences are to each other. For D segments, which are short and highly mutated, it may not be possible to find a confident alignment.
A contig is termed productive if the following conditions are met:
Full length requirement. The contig matches the initial part of a V gene. The contig continues on, ultimately matching the terminal part of a J gene.
Start requirement. The initial part of the V matches a start codon on the contig. Note that in the human and mouse reference sequences supplied by 10x Genomics, every V segment begins with a start codon.
Nonstop requirement. There is no stop codon between the V start and the J stop.
In-frame requirement. The J stop minus the V start equals one mod three. This just says that the codons on the V and J segments are in frame.
CDR3 requirement. There is an annotated CDR3 sequence (see below).
Structure requirement. Let VJ denote the sum of the lengths of the V and J segments. Let len denote the J stop minus the V start, measured on the contig. Then VJ - len lies between -25 and +25, except for IGH, which must be between -55 and +25. This condition is imposed to preclude anomalous structure changes that are unlikely to correspond to functional proteins.
For each contig, Cell Ranger searches for a CDR3 sequence using the conserved sequence that flanks the CDR3 region. Then the CDR3 sequence and its flanking regions are compared to motifs derived from V and J reference segments for human and mouse, as shown below. A letter represents a specific amino acid and a dot represents any amino acid:
left flank CDR3 right flank LQPEDSAVYY C... LTFG.GTRVTV VEASQTGTYF LIWG.GSKLSI ATSGQASLYL
Cell Ranger requires that a CDR3 sequence have at least 5 amino acids, start with a C, and not contain a stop codon. The flanking sequences for a candidate CDR3 are matched against the above motifs, and scored +1 for each position that matches one of the entries in a column.
For example, LTY.... scores 2 for the first three amino acids in the right flank. L matches an entry in the first column, contributing 1 to the score. T matches an entry in the second column, contributing 1 to the score. Y does not match the third column, and does not contribute to the score.
For a candidate CDR3 to be declared a CDR3 sequence, it must score at least 10. In addition the left flank must contribute at least 3 and the right flank must contribute at least 4.
Next, Cell Ranger finds the implied stop position of the end of the V segment on the contig. The implied stop is the start position of the V segment on the contig plus the length of the V segment. The CDR3 sequence is required to start at most 10 bases before the stop, and at most 20 bases after the stop of the V. These conditions for finding an implied stop are not applied in the denovo case.
If there is more than one CDR3 sequence, Cell Ranger chooses the one with the highest score. If there is a tie, the one with the later start position on the contig is chosen. If a tie remains, the longer CDR3 is chosen.
Cell Ranger 5.0+ annotates T cells as likely iNKT or MAIT cells based on the TCR V genes, J genes, and CDR3 sequences of the TCR alpha and TCR beta. For more information about iNKT and MAIT cells and how the annotation is performed, please see the iNKT and MAIT cell Algorithms documentation.