Cell Ranger7.1, printed on 04/20/2025
Cell Ranger is a set of analysis pipelines that process Chromium single cell data to align reads, generate feature-barcode matrices, perform clustering and other secondary analysis, and more (see list of example workflows and supported libraries). Cell Ranger includes five pipelines relevant to the 3' Single Cell Gene Expression Solutions and related products:
cellranger mkfastq demultiplexes raw base call (BCL) files generated by Illumina sequencers into FASTQ files. It is a wrapper around Illumina's bcl2fastq, with additional features that are specific to 10x Genomics libraries and a simplified sample sheet format.
cellranger count takes FASTQ files from cellranger mkfastq and performs alignment, filtering, barcode counting, and UMI counting. It uses the Chromium cellular barcodes to generate feature-barcode matrices, determine clusters, and perform gene expression analysis. The count pipeline can take input from multiple sequencing runs on the same GEM well. cellranger count also processes Feature Barcode data alongside Gene Expression reads.
cellranger multi is used to analyze Cell Multiplexing and Fixed RNA Profiling data. It takes FASTQ files from cellranger mkfastq and performs alignment, filtering, barcode counting, and UMI counting. It uses the Chromium cellular barcodes to generate feature-barcode matrices, determine clusters, and perform gene expression analysis. The cellranger multi pipeline also supports the analysis of Feature Barcode data.
cellranger aggr aggregates outputs from multiple runs of cellranger count or cellranger multi, normalizing those runs to the same sequencing depth and then recomputing the feature-barcode matrices and analysis on the combined data. The aggr pipeline can be used to combine data from multiple samples into an experiment-wide feature-barcode matrix and analysis.
cellranger reanalyze takes feature-barcode matrices produced by cellranger count, cellranger multi, or cellranger aggr and reruns the dimensionality reduction, clustering, and gene expression algorithms using tunable parameter settings.
Skip Cell Ranger download and installation and get started with 10x Genomics Cloud Analysis, our recommended method for running Cell Ranger pipelines for most new customers. Use your web browser to easily generate Cell Ranger outputs from your FASTQ files and aggregate outputs from multiple runs, free for every 10x Genomics sample. Currently available only in the United States and Canada. Sign up for a free account or view tutorials and learn more.
Learn how to install and run Cell Ranger.
If you are beginning with raw base call (BCL) files, the Cell Ranger workflow starts with demultiplexing the BCL files for each flow cell directory. 10x Genomics recommends using cellranger mkfastq as described in Generating FASTQs. If you are beginning with FASTQ files that have already been demultiplexed with bcl2fastq or bcl-convert directly, or from a public source such as SRA, you can skip cellranger mkfastq and begin with cellranger count. Please see the Specifying Input FASTQ pages (count, multi) for specific guidelines on which arguments to use for your scenario.
The exact steps of the workflow vary depending on how many samples, GEM wells, and flow cells you have, and whether you are including data from Feature Barcode, Cell Multiplexing, or Fixed RNA Profiling kits. This section describes a few possible workflows.
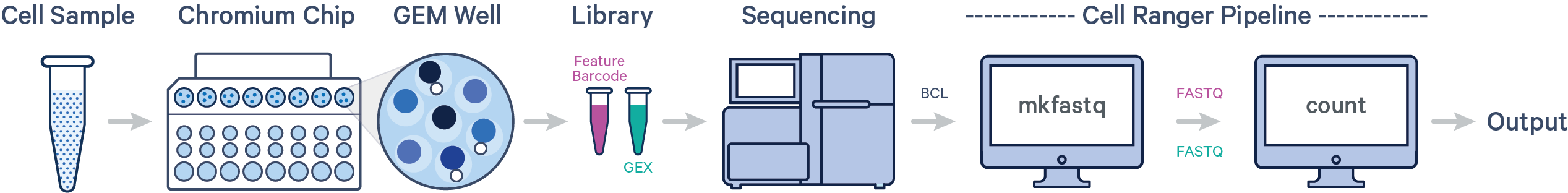
In this example, one sample is processed through one GEM well and sequenced on one flow cell. In this case, generate FASTQs using cellranger mkfastq and run cellranger count as described in Single-Sample Analysis.
This example also illustrates two sequencing libraries. A single GEM well can yield multiple physical libraries: one Gene Expression library and one or more Feature Barcode libraries.
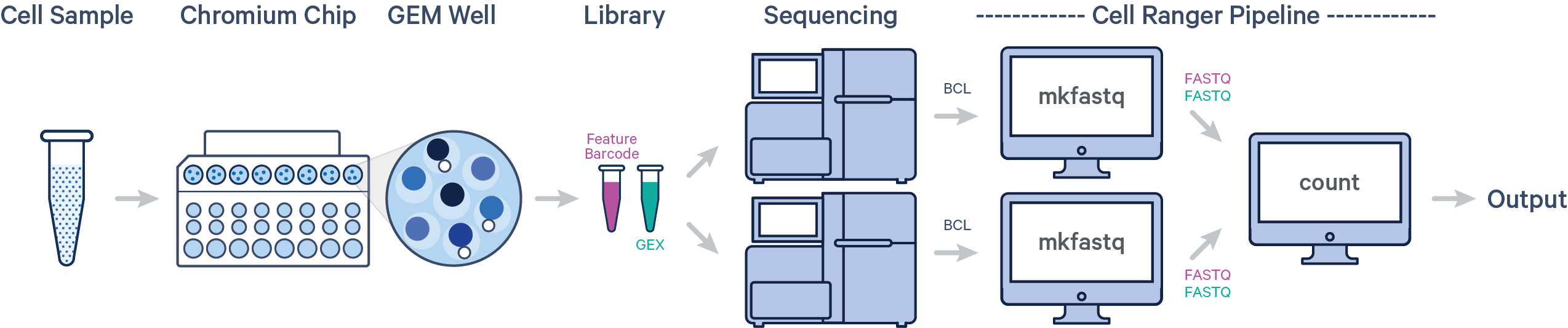
In this example, one sample is processed through one GEM well, resulting in one library which is sequenced across multiple flow cells. This workflow is commonly performed to increase sequencing depth. In this case, all reads can be combined in a single instance of the cellranger count or multi pipeline. This process is described in Specifying Input FASTQ pages (count, multi).
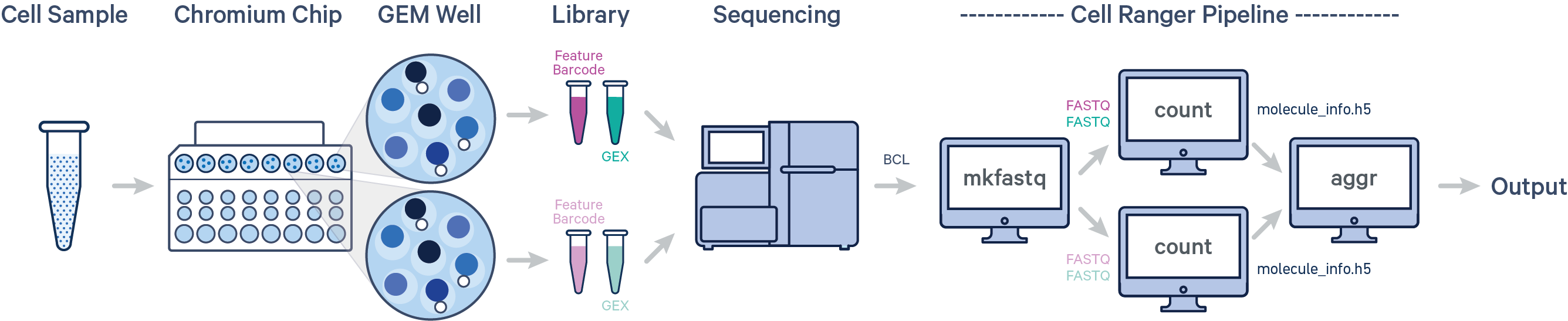
Here, one sample is processed through multiple GEM wells. This is typically done when conducting technical replicate experiments. The libraries from the GEM wells are then pooled onto one flow cell and sequenced. In this case, demultiplex the data from the sequencing run with cellranger mkfastq, then run the libraries from each GEM well through a separate instance of cellranger count. Then you can perform a combined analysis using cellranger aggr, as described in Multi-Library Aggregation.
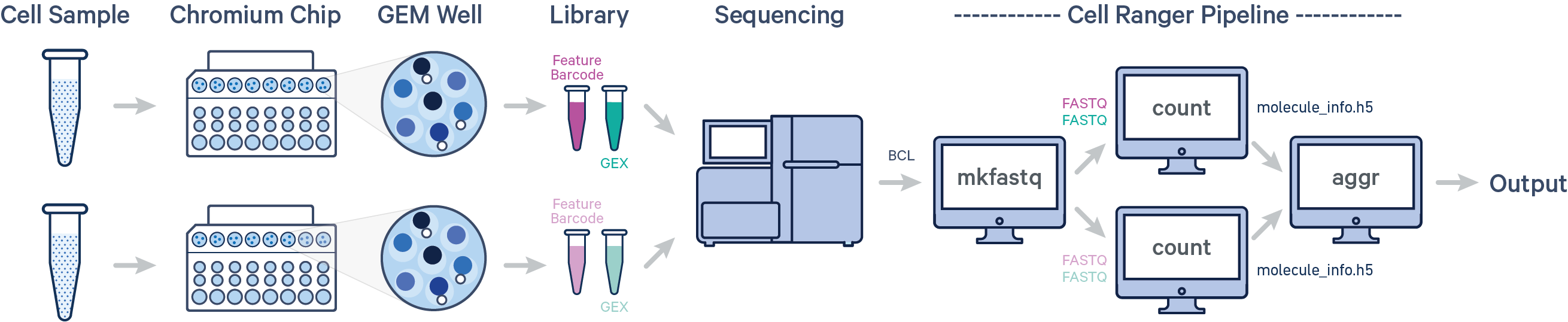
In this example, multiple samples are processed through multiple GEM wells, which generate multiple libraries and are pooled onto one flow cell. After demultiplexing, you must run cellranger count separately for each GEM well; if you have two GEM wells, then run cellranger count twice. Then you can aggregate them with a single instance of cellranger aggr, as described in Multi-Library Aggregation.
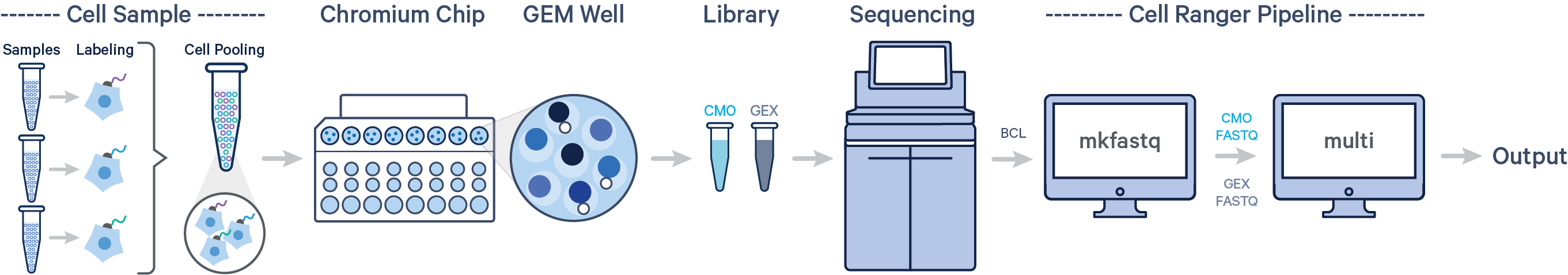
Cell Ranger 6.0 introduces support for analyzing Cell Multiplexing data. In this case, multiple samples are uniquely tagged with Cell Multiplexing Oligos (CMOs), enabling multiple samples to be pooled in a single GEM well. This results in a CMO and Gene Expression (GEX) library for each GEM well. After running cellranger mkfastq to generate FASTQ files, run the cellranger multi pipeline on the combined FASTQ data for the GEX and CMO libraries.
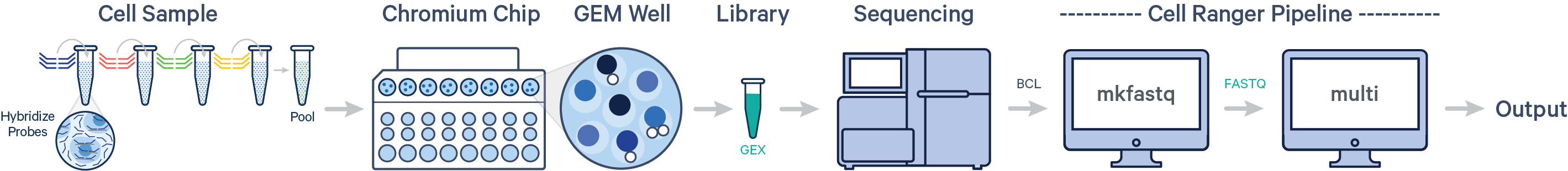
Cell Ranger 7.0 introduces support for analyzing Fixed RNA Profiling (FRP) Gene Expression data. In this case, multiple samples are uniquely tagged with Probe Barcodes, enabling samples to be pooled in a single GEM well and resulting in a Gene Expression library. After running cellranger mkfastq to generate FASTQ files, run the cellranger multi pipeline on the FASTQ data for the GEX library.
The library support of Cell Ranger 7.1 and previous versions is summarized in the tables below.
| Single Cell Gene Expression Solution | CR 7.1 | CR 7.0 | CR 6.1 | CR 6.0 | CR 5.0 | CR 4.0 | CR 3.1 | CR 3.0 | CR 2.2 |
|---|---|---|---|---|---|---|---|---|---|
| 3’ Gene Expression v2 Libraries | |||||||||
| 3’ Gene Expression v3 Libraries | |||||||||
| 3’ Gene Expression v3 + Cell Surface Protein Libraries | |||||||||
| 3’ Gene Expression v3 + CRISPR Screening Libraries | |||||||||
| 3’ Cell Surface Protein Libraries only | |||||||||
| Targeted Gene Expression | |||||||||
| 3’ Cell Multiplexing | |||||||||
| 3’ LT (Low Throughput) | |||||||||
| 3’ HT (High Throughput) | |||||||||
| Fixed RNA Profiling |