Cell Ranger3.0, printed on 03/29/2025
Cell Ranger is a set of analysis pipelines that process Chromium single-cell RNA-seq output to align reads, generate feature-barcode matrices and perform clustering and gene expression analysis. Cell Ranger includes four pipelines relevant to single-cell gene expression experiments:
cellranger mkfastq demultiplexes raw base call (BCL) files generated by Illumina sequencers into FASTQ files. It is a wrapper around Illumina's bcl2fastq, with additional useful features that are specific to 10x libraries and a simplified sample sheet format.
cellranger count takes FASTQ files from cellranger mkfastq and performs alignment, filtering, barcode counting, and UMI counting. It uses the Chromium cellular barcodes to generate feature-barcode matrices, determine clusters, and perform gene expression analysis. The count pipeline can take input from multiple sequencing runs on the same GEM well. cellranger count also processes Feature Barcoding data alongside Gene Expression reads.
cellranger aggr aggregates outputs from multiple runs of cellranger count, normalizing those runs to the same sequencing depth and then recomputing the feature-barcode matrices and analysis on the combined data. The aggr pipeline can be used to combine data from multiple samples into an experiment-wide feature-barcode matrix and analysis.
cellranger reanalyze takes feature-barcode matrices produced by cellranger count or cellranger aggr and reruns the dimensionality reduction, clustering, and gene expression algorithms using tunable parameter settings.
These pipelines combine Chromium-specific algorithms with the widely used RNA-seq aligner STAR. Output is delivered in standard BAM, MEX, CSV, HDF5 and HTML formats that are augmented with cellular information.
If you are beginning with raw base call (BCL) files, the Cell Ranger workflow starts with demultiplexing the BCL files for each flowcell directory. 10x recommends using cellranger mkfastq as described in Generating FASTQs. If you are beginning with FASTQ files that have already been demultiplexed with bcl2fastq directly, or from a public source such as SRA, you can skip cellranger mkfastq and begin with cellranger count. Please see the Specifying Input FASTQs page for specific guidelines on which arguments to use for your scenario.
The exact steps of the workflow vary depending on how many samples, GEM wells, and flowcells you have. This section describes the different possible workflows.
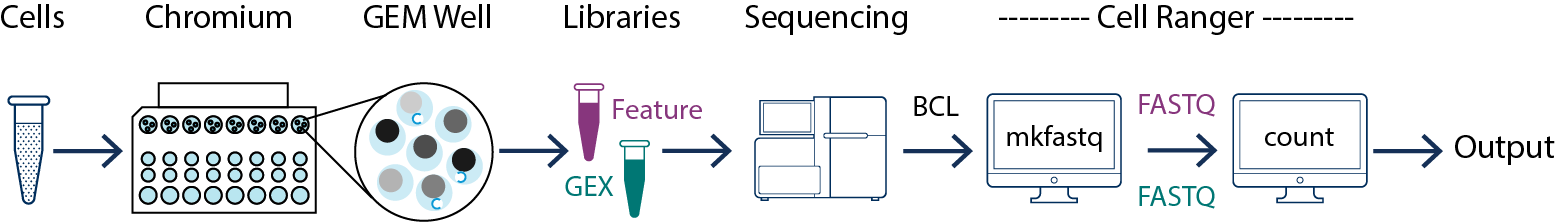
In this example you have one sample that is processed through one GEM well (a set of partitioned cells from a single 10x Chromium™ Chip channel) and sequenced on one flowcell. In this case you would generate FASTQs using cellranger mkfastq, and run cellranger count as described in Single-Sample Analysis.
This example also illustrates two sequencing libraries. A single GEM well can yield multiple libraries: one Gene Expression library, and one or more Feature Barcoding libraries.
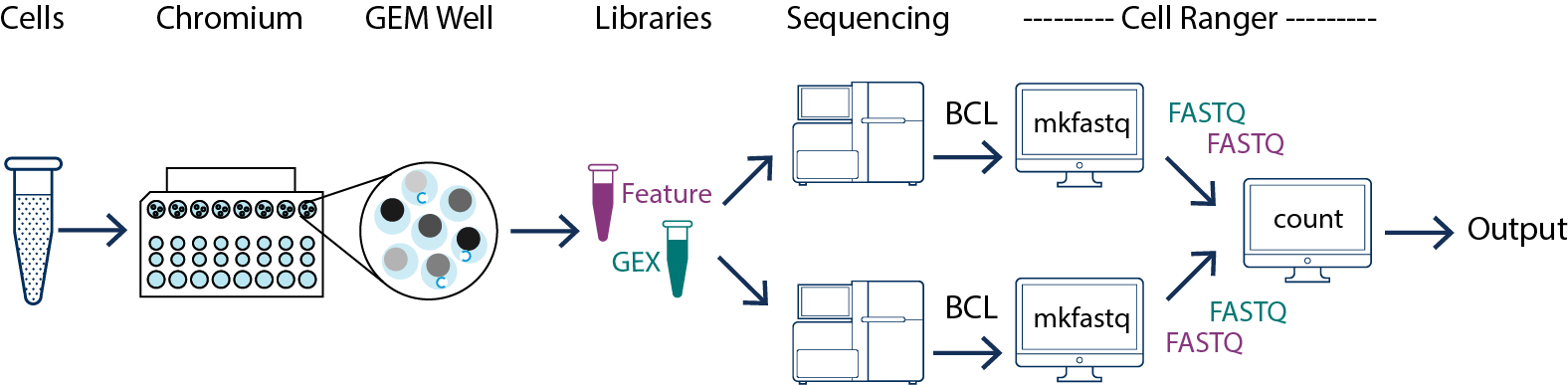
In this example you have one sample that is processed through one GEM well then you generate one library which is sequenced across multiple flowcells. This may be done to increase sequencing depth, for example. In this case all of the reads can be combined in a single instance of the cellranger count pipeline. This process is described in Specifying Input Fastqs.
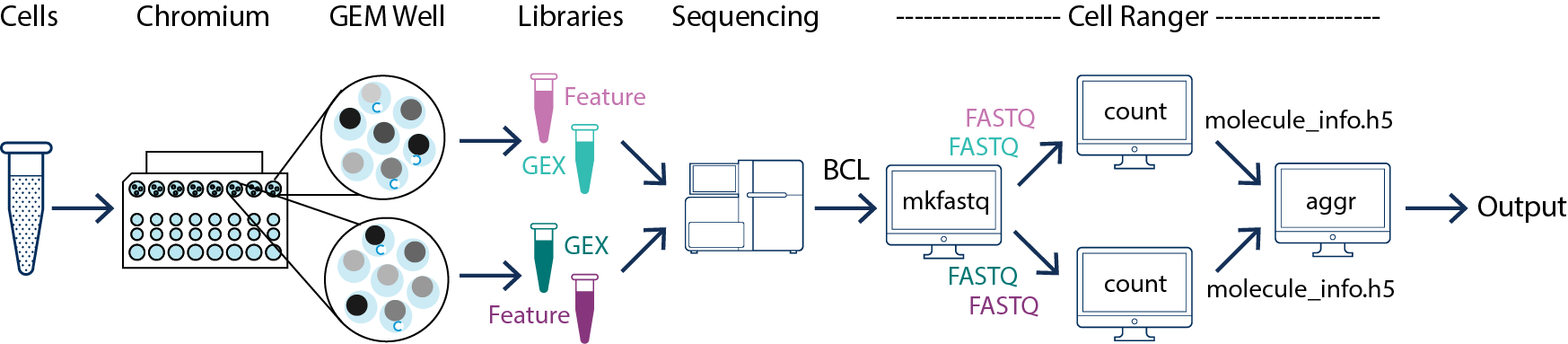
In this example you have one sample that is processed through multiple GEM wells. This is often done when conducting technical replicate experiments. The libraries from the GEM wells are then pooled onto one flowcell and sequenced. In this case you demultiplex the data from the sequencing run and then run the libraries from each GEM well through a separate instance of cellranger count. Once those are completed, you can perform a combined analysis using cellranger aggr, as described in Multi-Library Aggregation. (See figure above.)
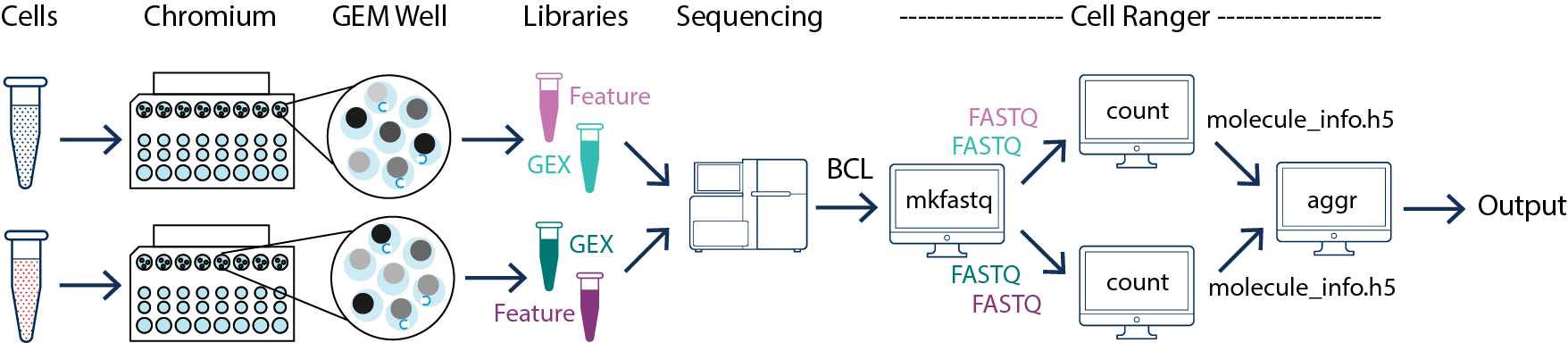
In this example you have multiple samples that are processed through multiple GEM wells which generate multiple libraries and are pooled onto one flowcell. In this case, after demultiplexing, you must run cellranger count separately for each GEM well. For instance, if your experiment involves two samples then you will have to run cellranger count two times. Then you can aggregate them with a single instance of cellranger aggr, as described in Multi-Library Aggregation.
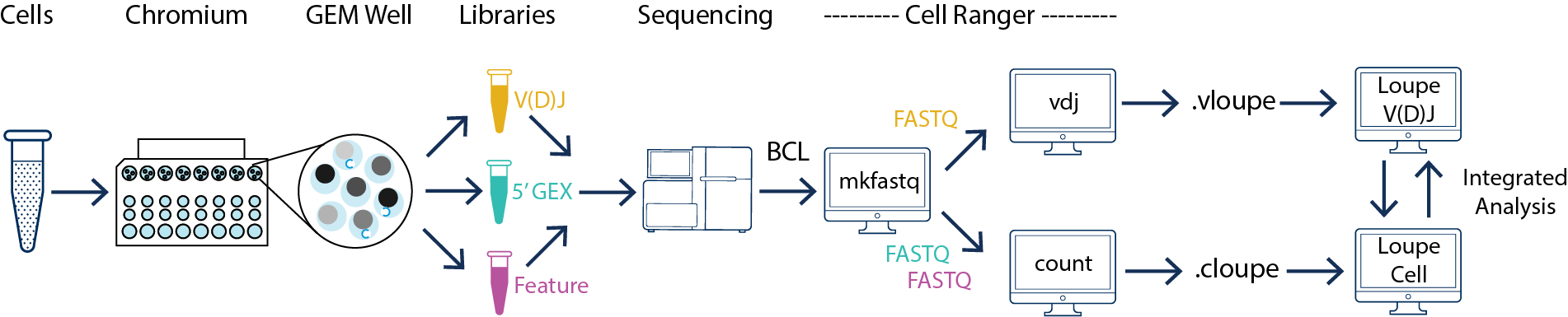
5′ gene expression libraries and V(D)J enriched libraries generated from the same cDNA product must be processed by cellranger count and cellranger vdj respectively. The outputs can be analyzed interactively using Loupe Cell Browser and Loupe V(D)J Browser. Please refer to Single Cell V(D)J + 5′ Gene Expression for more information.
Cell Ranger 3.0 is required for processing Single Cell 3' v3 libraries, or Feature Barcoding libraries. The assay support of Cell Ranger 3.0, and the previous Cell Ranger 2.2 is summarized below.
| Assay Type | Cell Ranger 3.0 | Cell Ranger 2.2 |
|---|---|---|
| Single Cell 3' v3 | Yes | No |
| Single Cell 3' v3 + Feature Barcoding | Yes | No |
| Single Cell 5' | Yes | Yes |
| Single Cell 5' + Feature Barcoding | Yes | No |
| Single Cell 3' v2 | Yes | Yes |
| Single Cell 3' v2 + Feature Barcoding | Yes | No |