Cell Ranger2.0, printed on 03/29/2025
Cell Ranger is a set of analysis pipelines that process Chromium single cell 3’ RNA-seq output to align reads, generate gene-cell matrices and perform clustering and gene expression analysis.
Cell Ranger 1.2 and later support libraries generated by the Chromium Single Cell 3' v1 and v2 reagent kits, whereas Cell Ranger 1.1 and earlier do not support v2 libraries.
Cell Ranger includes four main gene expression pipelines:
cellranger mkfastq wraps Illumina's bcl2fastq to correctly demultiplex Chromium-prepared sequencing samples and to convert barcode and read data to FASTQ files.
cellranger count takes FASTQ files from cellranger mkfastq and performs alignment, filtering, and UMI counting. It uses the Chromium cellular barcodes to generate gene-barcode matrices and perform clustering and gene expression analysis. count can take input from multiple sequencing runs on the same library.
cellranger aggr aggregates outputs from multiple runs of cellranger count, normalizing those runs to the same sequencing depth and then recomputing the gene-barcode matrices and analysis on the combined data. aggr can be used to combine data from multiple samples into an experiment-wide gene-barcode matrix and analysis.
cellranger reanalyze takes gene-barcode matrices produced by cellranger count or cellranger aggr and reruns the dimensionality reduction, clustering, and gene expression algorithms using tunable parameter settings.
These pipelines combine Chromium-specific algorithms with the widely used RNA-seq aligner STAR. Output is delivered in standard BAM, MEX, CSV, HDF5 and HTML formats that are augmented with cellular information.
Throughout the documentation, you will see references to samples, libraries and sequencing runs. We define these as follows:
The relationship between these terms can be complex:
The Cell Ranger workflow always starts with running cellranger mkfastq on each flowcell directory, as described in Generating FASTQs. The subsequent steps vary depending on how many samples, libraries and flowcells you have. We will describe them in order of increasing complexity:
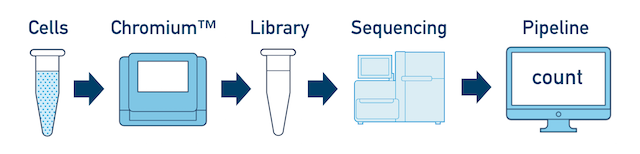
This is the most basic case. You have a single biological sample, which was prepared into a single library, and then sequenced on a single flowcell. Assuming the FASTQs have been generated with cellranger mkfastq, you just need to run cellranger count as described in Single-Library Analysis.
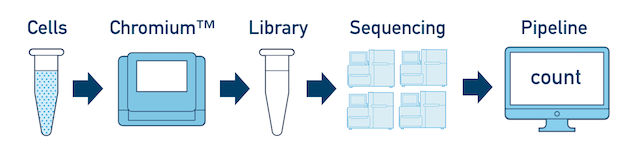
If you have a library which was sequenced across multiple flowcells, you can pool the reads from both sequencing runs. Follow the steps in Running Multi-Library Samples to combine them in a single cellranger count run.
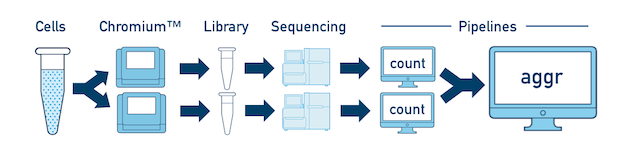
This comprises two scenarios.
If you prepared multiple libraries from the same sample (technical replicates, for example), then each one should be run through a separate instance of cellranger count. Once those are completed, you can perform a combined analysis using cellranger aggr, as described in Multi-Library Aggregation. (This is illustrated in the figure).
If you prepared multiple libraries from the same sample and want to pool them and analyze the combined data as a single sample, then you will need to use the MRO syntax to treat the multiple libraries as coming from a single sample. (This is not illustrated in the figure).
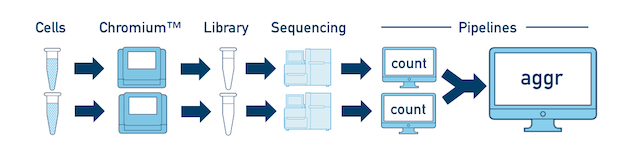
For a full experiment involving multiple biological samples, you must run cellranger count separately for each individual library deriving from each of those samples. For instance, if your experiment involves four samples, each having two libraries / replicates, then you will have to run cellranger count eight times. Then you can combine them all in a single call to cellranger aggr.