Cell Ranger3.0, printed on 04/02/2025
You have now seen how the chain panel allows you to explore and validate consensus sequences at a contig-by-contig level. If you have access to the contig BAM file generated by the Cell Ranger pipeline, you can further interrogate contigs of interest by exploring the individual reads that support a single contig alignment.
In this section, we're going to verify read support for the same single-base variant we investigated during our tour of the Sequence View. If you are coming directly from the previous page, you can skip directly to the next section: "Enabling Read Support for a .vloupe File".
If you're coming to this page from elsewhere, choose CDR3 Amino as the filter type below the clonotype list, and enter CAVSPNSGYALNF as the CDR3 sequence. Select the highlighted chain.
Next, in the chain panel, select Align Contigs and Consensus to Reference to see how the contigs and consensus sequence differ from the gene annotations in the reference. Note the SNP (mismatch) about halfway through the V gene in the consensus and contigs. Zoom into the base-level view by clicking on the mismatch, marked in orange on the contigs and consensus. You should see the chain sequence view as shown below:
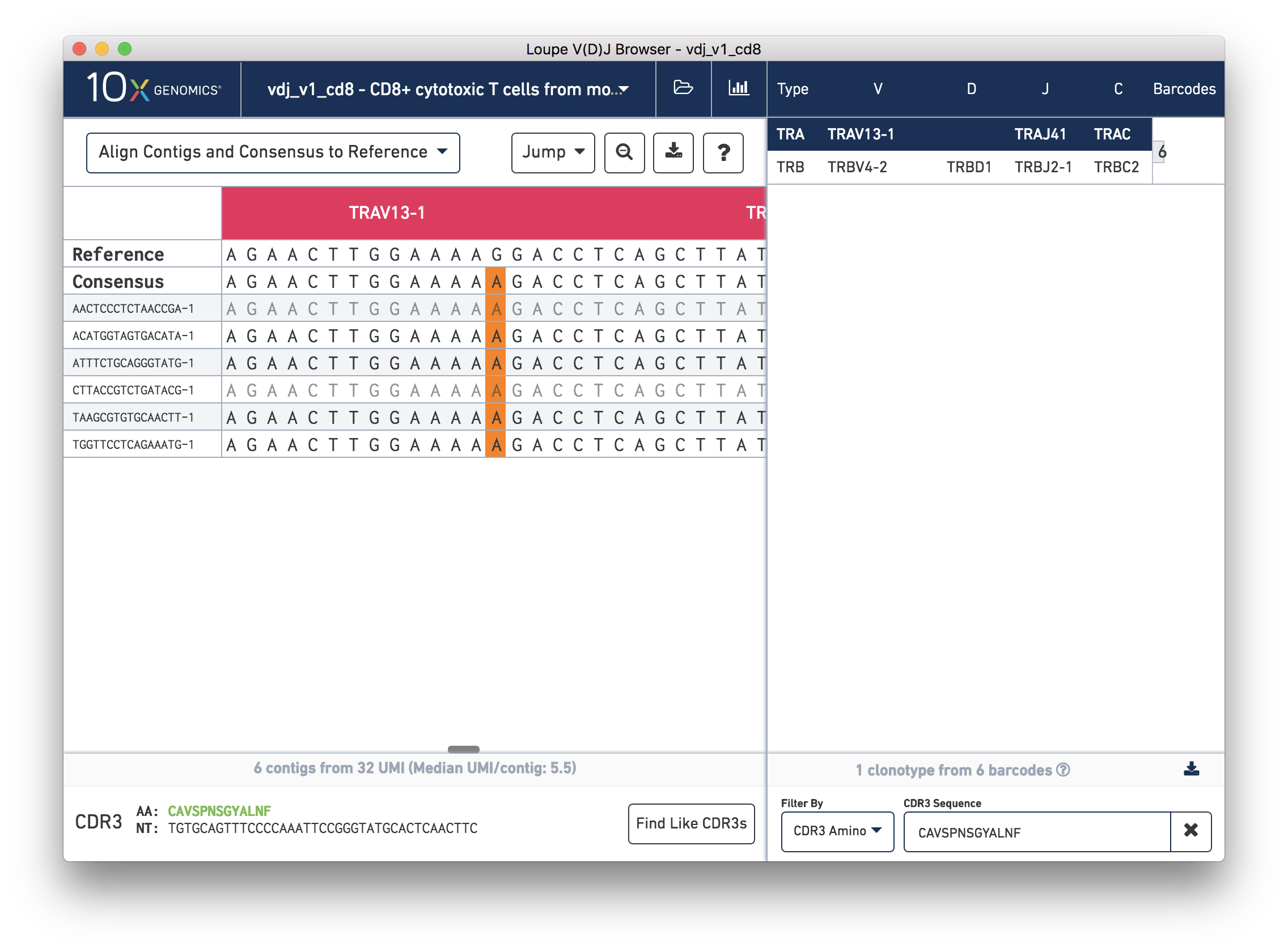
How confident should you be in that mismatch? Read support will tell us.
To view the supporting reads for any contig, hover over the names of the cell barcodes on the left side of the chain panel. For example, hover over the ATTTCTGCAGGGTATG-1 barcode, which should be the third contig for this chain. Once the popup appears, move your mouse into the popup, and press the Read Support button. You will be prompted, as shown below:
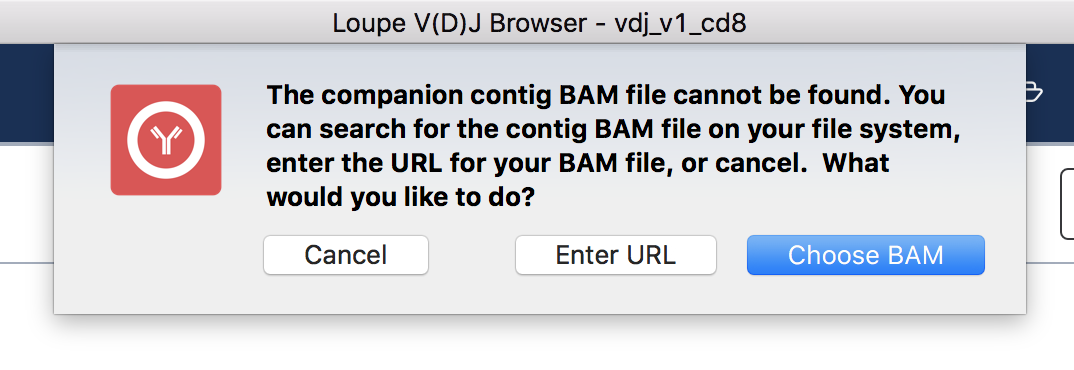
If the contig BAM file (all_contig.bam) is in the same folder as the corresponding .vloupe file, you won't see this message. Otherwise, you'll need to specify the path of the BAM file in your filesystem by selecting Choose BAM, or supply a web address where the BAM file can be found. In this example, we'll do the latter. Press Enter URL to enter an address for the BAM file.
The contig BAM file for the tutorial dataset can be found at
https://cg.10xgenomics.com/samples/cell-vdj/vdj_v1_cd8/vdj_v1_cd8_all_contig.bam.
Copy that address into the input box, and press Set URL.
| You can specify the contig BAM address for any 10x public dataset by going to the Datasets page, choosing a dataset, right-clicking the "Read-contig alignments" link from the list of output files, and copying the link address. Try it on the tutorial dataset! |
You should now see read support for the ATTTCTGCAGGGTATG-1_contig_1 contig, as shown below:
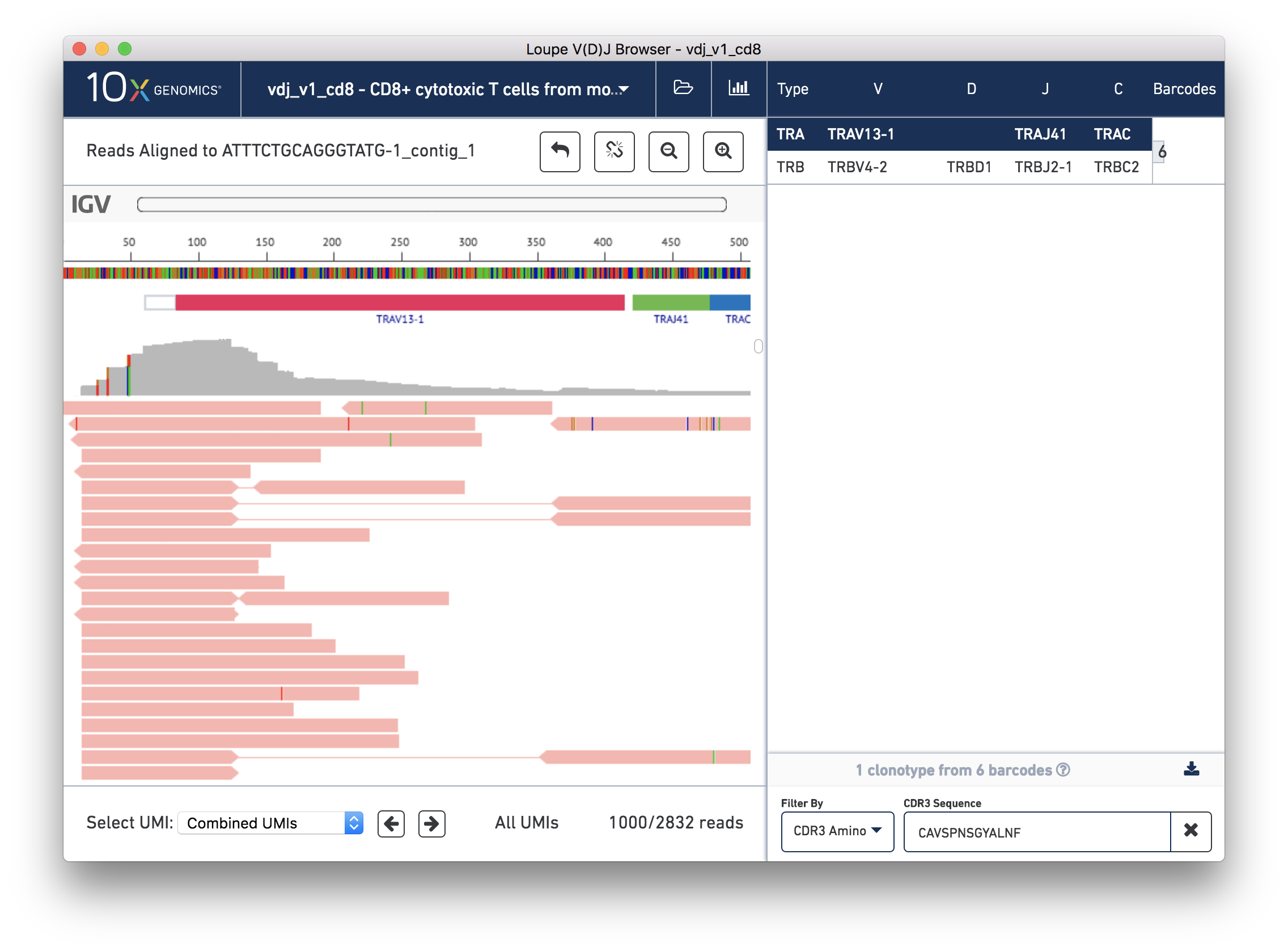
The individual reads are extracted from the BAM file and displayed by IGV.js, a lightweight version of the Integrative Genome Viewer, developed by the Broad Institute and University of California, San Diego. If you've used IGV, you should be familiar with the user interface. If not, the 'IGV User Interface' will offer a quick tour. Otherwise, you can skip ahead to Validating a SNP.
There are five distinct regions in the IGV read support view. In order from top to bottom, they are:
The zoom bar. When zoomed in, this bar will tell you where you are within the larger contig by highlighting the region being shown. You can also click in the zoom bar to recenter your viewport.
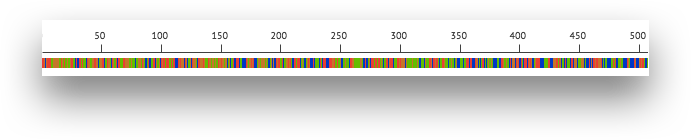
The ruler and base bar. The ruler measures the position of the base relative to the beginning of the contig. The sequence of the assembled contig is rendered below the ruler. When zoomed in such that space permits, this will show the single letter abbreviations for the nucleotides. At lower resolutions, it will show a color coded band to indicate the base: A is green, C is blue, G is orange and T is red.
The annotation bar. This shows the gene annotations along the contig. These should match the gene annotations from the chain view, and may be useful for undersstanding how the chain view relates to this read support view.

The coverage bar. This shows you a histogram of coverage per base across all reads, across all UMIs. If there is significant discordance in a particular position, you'll see a vertical color bar drawn at that spot (see the three above). Clicking on the bar will show you the breakdown in observed bases at that position. You should expect to see more reads at the 5′ end of the transcript.

Finally, the remainder of the IGV view shows the reads themselves. Reads are represented by thick horizontal bars. Paired (R1/R2) reads will be linked by a thin horizontal line. Reads are colored by UMI; if you scroll down using your mouse wheel or the scrollbar to the right of the reads, you will see where the reads from one amplified transcript end and reads from another transcript begin. Variants appear as colored bars superimposed on the read, including deletions, which appear as gaps. Clicking on a read will reveal its name, strand, cigar string, and other information encoded in BAM tags.
Below the IGV read support view is a panel by which you can navigate the list of reads by individual UMI. For performance reasons, we show a maximum of 1000 reads at a time.
Remember from the chain view that there was a single-base variant (G->A) roughly in the middle of the V gene annotation. This variant ends a 5-A long segment following a 'TTGG'. To check read support for this variant, click the Zoom In button three times, or enough times until you can see the base letters.
The 5-A string in question should be between bases 263 and 267 on the contig, inclusive. The variant in question is the last base in the string. To check how consistent that variant was within the reads, you can click the coverage bar directly below the 5th A. You may have to scroll up back to the coverage bar if you've scrolled down to view more reads.
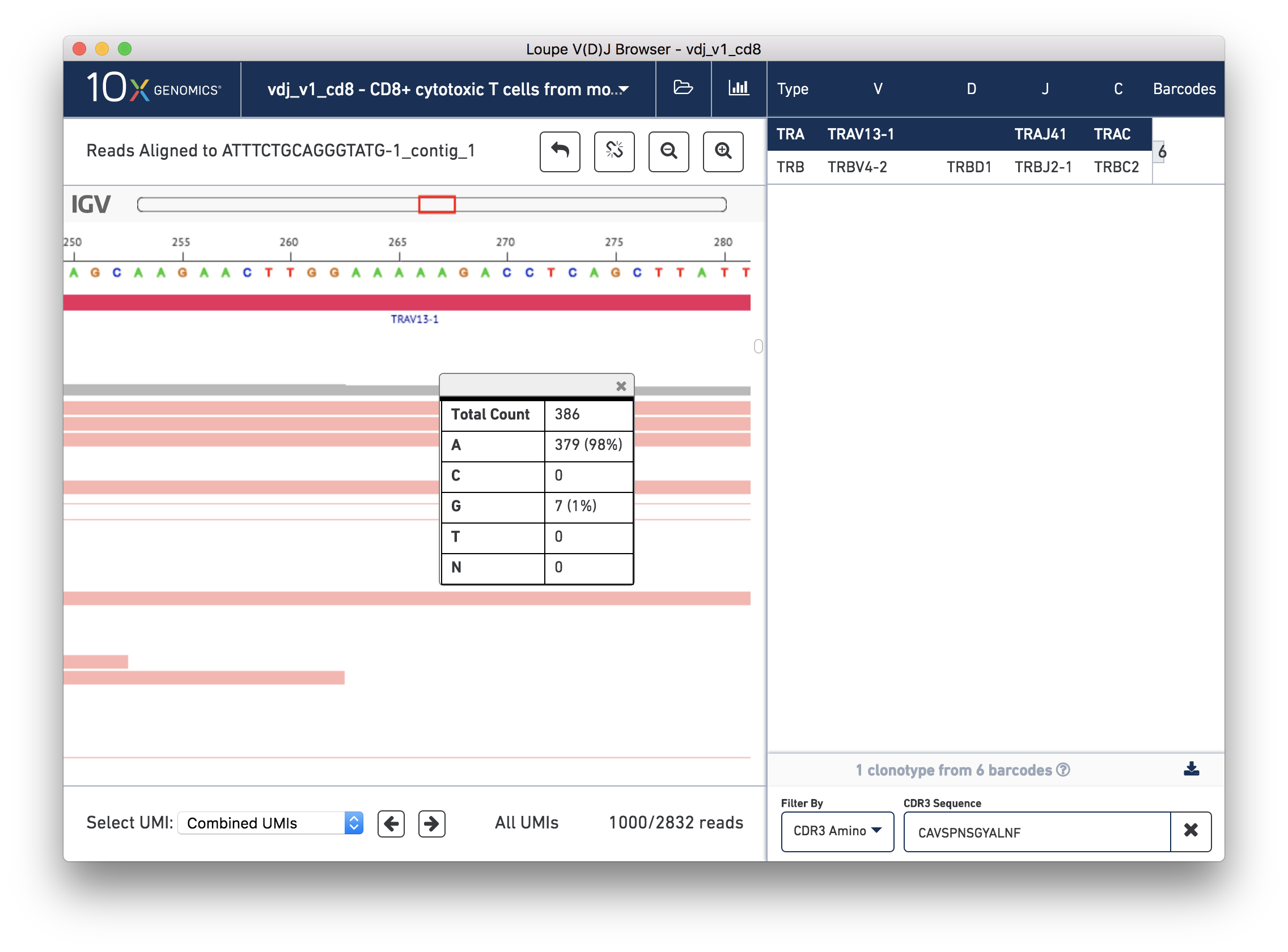
You can see that 98% of the reads that covered that base had an A, instead of the G as in the reference. Scrolling through the reads should give you additional confidence; there is little discordance in the base 267 spot for this contig.
Now that read support has confirmed the variant, we can observe how frequently this variant occurs within the sample. This variant occurs in the TRAV13-1 gene. In the Filter Type dropdown below the clonotype list, select Gene Name, and then enter TRAV13-1 in the input box. Click on each occurrence of TRAV13-1 that appears in the list. You'll note that some chains will include the SNP, and others will not. Roughly half of the chains have the variant, indicating a heterozygous allele (G|A) at that location.
Up to this point, we’ve been analyzing a T cell sample. The Chromium Single Cell V(D)J Solution also supports the sequencing and analysis of B cell immunoglobulins. Let’s download and analyze a B cell sample.