Cell Ranger7.1, printed on 04/15/2025
This page describes the output file structure from the cellranger multi subcommand specifically for 3' Cell Multiplexing data. This subcommand was introduced in Cell Ranger 5.0 for joint analysis of 5' gene expression and V(D)J (GEX + VDJ) data, and in Cell Ranger 6.0 for 3' Cell Multiplexing data.
Upon completion, the cellranger multi subcommand will produce an outs/ directory with the following structure:
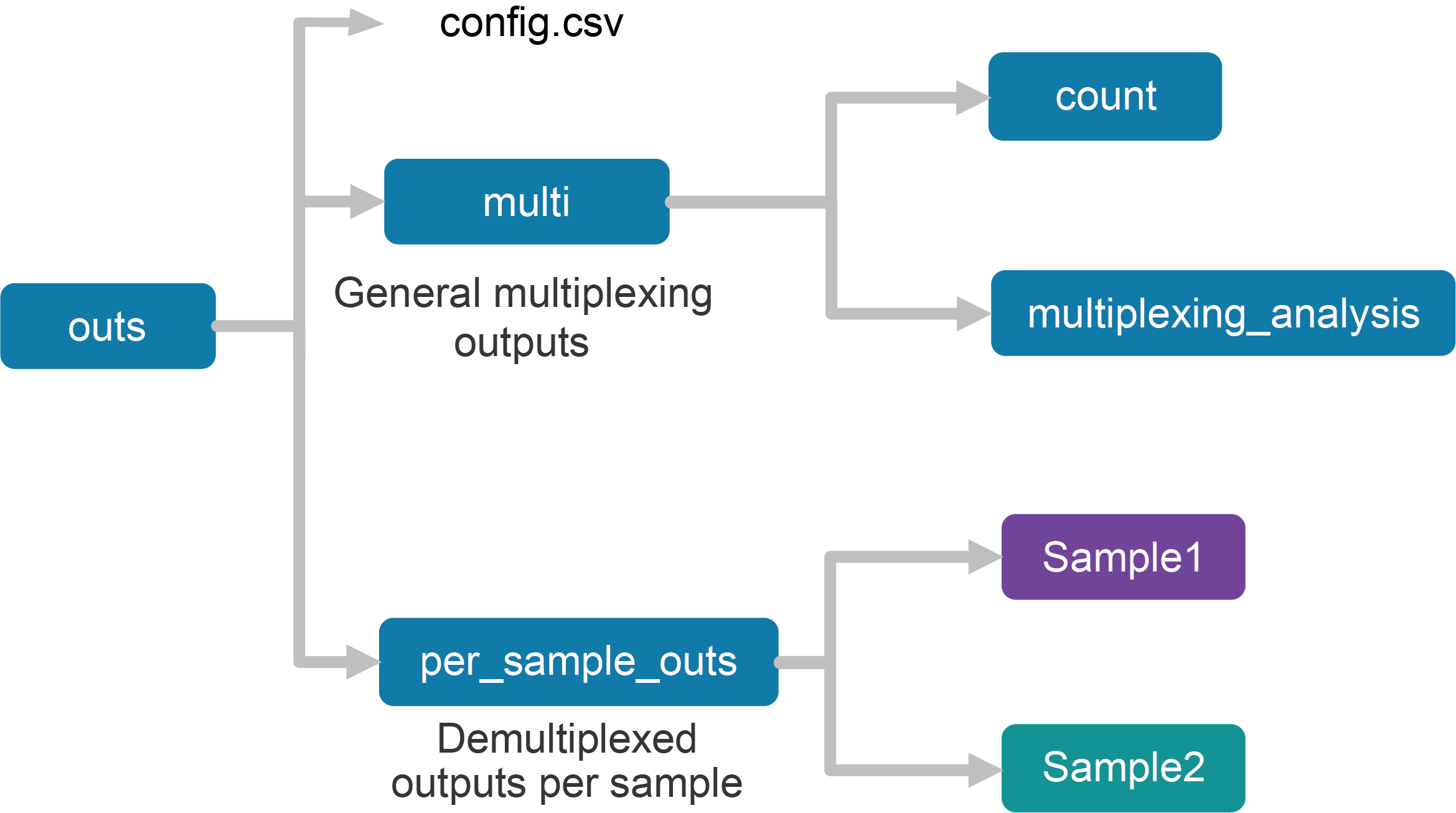
Using the tree Linux command, the file structure looks like this:
├── config.csv
├── multi
│ ├── count
│ └── multiplexing_analysis
└── per_sample_outs
├── Sample1
└── Sample2
The first section of the outputs contains the config.csv file, a duplicate of the input config CSV file. The files in the multi folder are generic to the entire Cell Multiplexing experiment, while the files in the per_sample_outs directory have been demultiplexed to single samples.
Within the multi directory, there are count and multiplexing_analysis directories:
└─ multi
├── count
│ ├── feature_reference.csv
│ ├── raw_cloupe.cloupe
│ ├── raw_feature_bc_matrix
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_feature_bc_matrix.h5
│ ├── raw_molecule_info.h5
│ ├── unassigned_alignments.bam
│ └── unassigned_alignments.bam.bai
└── multiplexing_analysis
├── assignment_confidence_table.csv
├── cells_per_tag.json
├── tag_calls_per_cell.csv
└── tag_calls_summary.csv
The count directory contains raw files that include cells and background data:
| Output File | Description |
|---|---|
feature_reference.csv |
Feature reference (contains both CMO and Feature Barcode) used for this sample |
raw_cloupe.cloupe |
A Loupe-readable file containing all cell-associated barcodes in the experiment. This cloupe file also contains UMI counts for all tags (prior to tag assignments), which could be useful for troubleshooting Cell Multiplexing library issues. |
raw_feature_bc_matrix |
A matrix of UMI counts associated with a feature (row) and a barcode (column), in MEX format, including both the GEX and CMO feature counts. This matrix contains every barcode from the fixed list of known good barcode sequences that has at least one read. This includes background and cell-associated barcodes. |
raw_feature_bc_matrix.h5 |
A matrix of UMI counts associated with a feature (row) and a barcode (column), in H5 format, including both the GEX and CMO feature counts. This matrix contains every barcode from the fixed list of known good barcode sequences that has at least one read. This includes background and cell-associated barcodes. |
raw_molecule_info.h5 |
Information about all molecules in the experiment. This file includes background and cell-associated barcodes, and cannot be used as input for cellranger aggr pipeline. |
unassigned_alignments.bam |
Alignments from barcodes not assigned to any sample. |
unassigned_alignments.bam.bai |
Alignments from barcodes not assigned to any sample (index). In cases where the reference transcriptome is generated from a genome with very long chromosomes (>512 Mbp), Cell Ranger v7.0+ generates an unassigned_alignments.bam.csi index file instead. |
The multiplexing_analysis directory contains:
| Output File | Description |
|---|---|
assignment_confidence_table.csv |
A table that contains all information from the tag assignment algorithm for each cell-associated barcode, in CSV format. More details below. |
cells_per_tag.json |
Lists the cell-associated barcodes that were assigned a given tag, for each tag, in JSON format. |
tag_calls_summary.csv |
A table that summarizes the multiplexing results, including the number of cells assigned no tag, one tag, and more than one tag, in CSV format. More details below. |
tag_calls_per_cell.csv |
This table summarizes the assigned tags and the UMI counts per tag for all barcodes assigned as singlets or multiplets in a CSV format. Barcodes called as Unassigned or Blanks are not included in this table. More details below. |
barcode_sample_assignments.csv |
If barcode-sample-assignment was specified in the multi config CSV, a duplicate of the input barcode-sample assignment CSV file will be generated. |
The assignment_confidence_table.csv table provides a summary of all the information from the tag-assignment algorithm for each cell-associated barcode, including the probability that a given barcode belongs to a given state. The user may modify the confidence threshold for assigning tags to barcodes on their own in a data-science environment like Python or R to enable further downstream analysis.
Row,CMO301,CMO302,Barcode,Multiplet,Blank,Assignment,Assignment_Probability,CMO301_cnts,CMO302_cnts 0,0.999721559521299,3.4331367105764475e-10,AAACCCAAGAGTGTGC-1,0.0002784107425317906,2.939285533268778e-08,CMO301,0.999721559521299,4.0744873049856905,2.444044795918076 1,2.1178952222935904e-09,0.9999759641250832,AAACCCAAGATGCTTC-1,2.3239920572975783e-06,2.1709764964310346e-05,CMO302,0.9999759641250832,2.6020599913279625,3.8196755199942927 2,0.999822627035718,3.287157579467574e-07,AAACCCAAGGTACAGC-1,8.871810165359213e-05,8.83261468704939e-05,CMO301,0.999822627035718,3.630834517828051,2.3404441148401185 3,0.8947739014808398,9.424785606470267e-09,AAACCCAAGTAGCTCT-1,0.10522608718358256,1.910792092383626e-09,Unassigned,0.8947739014808398,4.219767844658398,2.9916690073799486 4,0.033490762078236785,0.004751323355950833,AAACCCACACGGATCC-1,0.9617539697731167,3.944792695789878e-06,Multiplet,0.9617539697731167,3.6148972160331345,3.4896772916636984Column descriptions:
Column descriptions:
CMO301, CMO302, etc.: one column per tag used in the experiment, indicates the probability that a given barcode belongs to each of those singlet statesBarcodes: the cell-associated barcodeMultiplet: the coarse-grained multiplet state, which contains the probability that the given barcode is some-type of multiplet. Obtained by summing the probabilities across each possible multiplet state in the experimentBlanks: contains the probability that the barcode contains un-tagged cells i.e., is considered to have too few tag counts for every tagAssignment: the state assigned by the tag-assignment algorithm for the given barcode. Values: one of the singlet states, Multiplet, Blanks, or Unassigned (if every state has probability less than 90%, which is the default minimum confidence threshold.)Assignment_Probability: the probability of the most-likely stateCMO301_cnts, CMO302_cnts, etc.: (starting from Cell Ranger v7.1) one column per tag used in the experiment, indicates the log-transformed UMI count for each tagThe tag_calls_summary.csv summarizes multiplexing results by providing statistics about categories including the number of cells assigned no tag, one tag, more than one tag, etc. The category No tag assigned includes both cells that were considered Blanks and cells considered Unassigned.
Category,num_cells,pct_cells,median_umis,stddev_umis No tag molecules,0,0.0,None,None No tag assigned,386,2.9,None,None 1 tag assigned,12465,93.6,None,None More than 1 tag assigned,472,3.5,None,None CMO301,6437,48.3,3442.0,7988.3 CMO302,6028,45.2,3515.5,5167.2 CMO301|CMO302,472,3.5,12414.0,9696.0
Column descriptions:
num_cells: number of cells in the categorypct_cells: percent of cells in the categorymedian_umis: median UMI counts for the tag(s), amongst cells assigned those tag(s)stddev_umis: standard deviation of the UMI counts for the tag(s), amongst cells assigned those tag(s)The tag_calls_per_cell.csv file contains tag calls per cell, one line for each barcode. It contains all singlet and multiplet cells; Unassigned or Blanks are not included.
cell_barcode,num_features,feature_call,num_umis AAACCCAAGCAACAGC-1,1,CMO301,16778 AAACCCAAGCTCGTGC-1,1,CMO301,1735 AAACCCACATGACTGT-1,1,CMO301,1625 AAACCCAGTCCACAGC-1,1,CMO301,19323 AAACCCAGTCGCGGTT-1,1,CMO301,1678
Column descriptions:
cell_barcode: cell-barcodenum_features: number of tags assigned to that cellfeature_call: names of tag(s) assigned, delimited by "|"num_umis: number of molecules (UMIs) for each tag assigned, delimited by "|"The per_sample_outs directory contains sample-level files with data from cells only (background data filtered out):
├── count │ ├── analysis │ │ ├── clustering │ │ ├── diffexp │ │ ├── pca │ │ ├── tsne │ │ └── umap │ ├── sample_cloupe.cloupe │ ├── feature_reference.csv │ ├── sample_alignments.bam │ ├── sample_alignments.bam.bai │ ├── sample_filtered_barcodes.csv │ ├── sample_filtered_feature_bc_matrix │ │ ├── barcodes.tsv.gz │ │ ├── features.tsv.gz │ │ └── matrix.mtx.gz │ ├── sample_filtered_feature_bc_matrix.h5 │ └── sample_molecule_info.h5 ├── metrics_summary.csv └── web_summary.html
Note that the sample_filtered_feature_bc_matrix directory and sample_filtered_feature_bc_matrix.h5 file are similar to the filtered_feature_bc_matrix and filtered_feature_bc_matrix.h5, respectively, generated by cellranger count. For more information about these files, see the Feature Barcode matrices section. These are the key files that contain expression levels, which can be used for downstream analysis and data interpretation.
|
The per_sample_outs directory contains:
| Output File | Description |
|---|---|
count/ |
Folder containing the results of any gene expression and Feature Barcode analysis, see table below. |
metrics_summary.csv |
Run summary metrics file in CSV format, described in the Cell Multiplexing metrics page. |
web_summary.html |
Run summary metrics and charts in HTML format, described in the multi web summary page. |
The count directory contains:
| Output File | Description |
|---|---|
analysis/ |
Folder containing the results of graph-based clusters and K-means clustering 2-10; differential gene expression analysis between clusters; and PCA, t-SNE, and UMAP dimensionality reduction. Learn more |
sample_cloupe.cloupe |
A Loupe Browser visualization and analysis file, containing only cells assigned to this sample. |
feature_reference.csv |
A duplicate of the input Cell Multiplexing feature reference CSV file |
sample_alignments.bam |
Indexed BAM file containing position-sorted reads aligned to the genome and transcriptome, as well as unaligned reads, annotated with barcode information. Learn more |
sample_alignments.bam.bai |
Index file for the sample_alignments.bam. In cases where the reference transcriptome is generated from a genome with very long chromosomes (>512 Mbp), Cell Ranger v7.0+ generates a sample_alignments.bam.csi index file instead. |
sample_filtered_barcodes.csv |
File containing a list of only cell-associated barcodes. |
sample_filtered_feature_bc_matrix/ |
Contains only detected cell-associated barcodes in MEX format. Each element of the matrix is the number of UMIs associated with a feature (row) and a barcode (column), as described in the feature-barcode matrix page, including both the GEX and CMO feature counts. This file can be input into third-party packages and allows users to wrangle the feature-barcode matrix (e.g. to filter outlier cells, run dimensionality reduction, normalize gene expression). |
sample_filtered_feature_bc_matrix.h5 |
Same information as sample_filtered_feature_bc_matrix in HDF5 format, including both the GEX and CMO feature counts. |
sample_molecule_info.h5 |
Contains per-molecule information for all molecules that contain a valid barcode, valid UMI, and were assigned with high confidence to a gene or Feature Barcode. This file is a required input to run cellranger aggr. Learn more |
For Cell Multiplexing experiments with Antibody Capture libraries, there will be additional outputs. These files are described in the Antibody outputs overview. The output structure will include an additional antibody_analysis directory in the per_sample_outs/count directory:
├── count │ ├── analysis │ ├── sample_cloupe.cloupe │ ├── antibody_analysis │ │ └── aggregate_barcodes.csv │ ├── feature_reference.csv │ ├── sample_alignments.bam │ ├── sample_alignments.bam.bai │ ├── sample_filtered_barcodes.csv │ ├── sample_filtered_feature_bc_matrix │ ├── sample_filtered_feature_bc_matrix.h5 │ └── sample_molecule_info.h5 ├── metrics_summary.csv └── web_summary.html
For Cell Multiplexing experiments with CRISPR Guide Capture libraries, there will be additional outputs. These files are described in the CRISPR outputs overview. The output structure will include an additional crispr_analysis directory in the per_sample_outs/count directory:
├── count │ ├── analysis │ ├── sample_cloupe.cloupe │ ├── crispr_analysis │ │ ├── cells_per_protospacer.json │ │ ├── feature_reference.csv │ │ ├── perturbation_effects_by_feature │ │ ├── perturbation_effects_by_target │ │ ├── perturbation_efficiencies_by_feature.csv │ │ ├── perturbation_efficiencies_by_target.csv │ │ ├── protospacer_calls_per_cell.csv │ │ ├── protospacer_calls_summary.csv │ │ ├── protospacer_umi_thresholds.csv │ │ └── protospacer_umi_thresholds.json │ ├── feature_reference.csv │ ├── sample_alignments.bam │ ├── sample_alignments.bam.bai │ ├── sample_filtered_barcodes.csv │ ├── sample_filtered_feature_bc_matrix │ ├── sample_filtered_feature_bc_matrix.h5 │ └── sample_molecule_info.h5 ├── metrics_summary.csv └── web_summary.html