Cell Ranger6.0, printed on 03/11/2025
During the clonotype grouping stage, cell barcodes are placed in groups called clonotypes. Each clonotype consists of all descendants of a single, fully rearranged common ancestor, as approximated computationally. During this process, some cell barcodes are flagged as likely artifacts and filtered out, meaning that they are no longer called as cells.
T cells. The lack of somatic hypermutation in T cell receptors (TCRs) yields biological clonotypes which have identical V(D)J transcripts. Technical artifacts (e.g. arising in reverse transcription) can result in the computed clonotypes having isolated differences. These are generally rare.
B cells. Fully rearranged B cell receptors (BCRs) can undergo somatic hypermutation (SHM), which can increase antigen affinity. Thus for BCRs, V(D)J transcripts in a clonotype can differ at any position, as shown below:
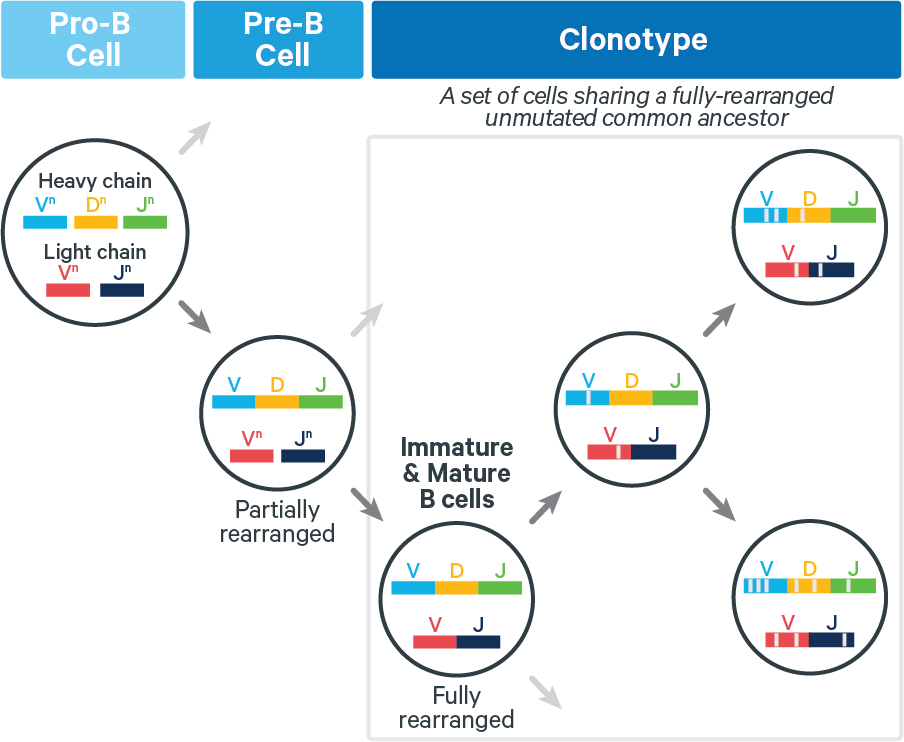
Because SHM can introduce numerous mutations, it is challenging to accurately infer B cell
clonotypes. Cell Ranger version 5.0 and onwards accomplishes this by invoking a module for clonal
analysis called enclone, which simultaneously groups cells into clonotypes and filters some out.
enclone is also available in beta as a separate command-line, fast, exploratory tool,
see https://10xgenomics.github.io/enclone or
bit.ly/enclone. The command
line form has arguments permitting granular control over the clonotyping and filtering
heuristics. enclone can also display clonotypes and infer phylogenetic trees. enclone as a
command-line tool is frequently updated and supported via
[email protected], separately from Cell Ranger.
Clonotyping grouping and the associated filtering are complex and heuristic. The concepts are described briefly here, and some link out to the enclone site for technical documentation.
A. Clonotype grouping. This includes two key steps:
Step 1: Cells are placed into groupings called exact subclonotypes if they have essentially identical V(D)J transcripts, meaning the sequences extending from the start codon through the J gene are identical, and the constant region gene assignments are the same. The algorithm does not test for somatic hypermutations (SHM) in the 5’-UTR or constant region, and moreover the data normally extend only up to the primer site in the constant region.
Step 2: Only productive transcripts are used. Cells which are missing a chain (typically because of lower coverage) are placed in their own exact subclonotype, although they may later be joined into the same clonotype (below).
Exact clonotypes are iteratively merged into clonotypes based on comparing each pair of exact subclonotypes to each other. They can be merged into a single clonotype based on two types of evidence:
(a) Similarity of their rearrangement junction regions. Somatic hypermutation during clonal evolution can cause junction regions to diverge.
(b) Evidence of shared SHM outside their junction regions. This is assessed by identifying positions within V genes that are identical on the two exact subclonotypes, differ from the reference, and do not represent germline differences. An outline of how this is implemented is described on the enclone site.
B. Cell filtering during the clonotype grouping process. During library generation, artifacts can arise by two primary mechanisms:
(a) Reverse transcription or sequencing can introduce base call errors. These usually occur at bases having low quality scores. Cells with these low quality bases are screened out, typically at a low rate.
(b) GEMs may contain material from two or more cells: either entire intact cells, cell fragments, or individual mRNA molecules. Detection of contamination is complex and is accomplished via multiple heuristic filters. A partial guide to the filters can be found on the enclone website. Note that if the multi pipeline is run with both gene expression and V(D)J data, then barcodes which are not called cells by the gene expression pipeline will also be deleted from the V(D)J cell set.