Cell Ranger ATAC2.0, printed on 04/02/2025
The computational pipeline for processing the data produced from the Chromium Single Cell ATAC Solution involves the following analysis steps:
We first detect the location of the 10x barcode within the i5 index read: the first 16 bases or the last 16 bases and in either the forward or reverse orientation. We pick the location that maximizes the fraction of barcodes that are valid. Once the location is determined we perform Hamming distance 1 error correction to account for sequencing error. Each barcode sequence is checked against an allowed list of valid barcode sequences, and the frequency of each valid barcode is counted. We attempt to error correct invalid barcodes that are not on the allowed list, by finding all valid barcodes within 1 mismatch of the observed sequence, and scoring them based on the abundance of that barcode in the read data and quality value of the incorrect bases. An observed invalid barcode is corrected if it has > 90% probability of being the real barcode based on this model.
Cell Ranger ATAC performs reference-based analysis and requires adapter and primer oligo sequence to be trimmed off before mapping confidently. In the current chemistry, the 3' end of a read (the end of the read sequence) may contain the reverse complement of the primer sequence, if the read length is greater than the length of the genomic fragment. We use an algorithm that is similar to the cutadapt tool to identify the reverse complement of the primer sequence at the end of each read, and trim it from the read prior to alignment.
Then, the trimmed read-pairs are aligned to a specified reference using a modified version of the BWA-MEM algorithm. We align read pairs using a fixed prior on the insert size distribution, which is assumed to be gaussian with a mean of 250 and standard deviation of 150. BWA-MEM divides read pairs into batches and determines this number on the fly. The results of either approach are very similar especially for high MAPQ read pairs that are used in downstream analyses.
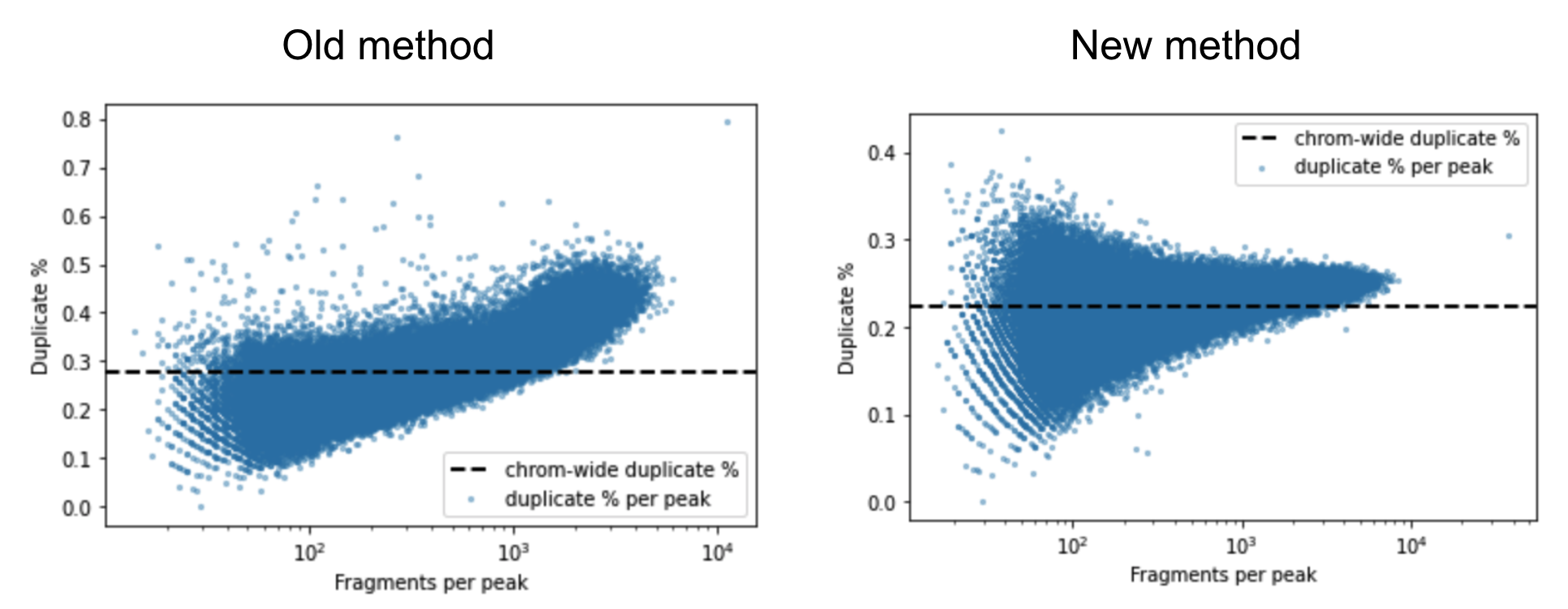
A barcoded fragment that represents a template molecule is amplified during the library preparation process and this results in multiple read pairs being sequenced that arise from the same original template molecule. One of these read pairs is selected to represent the template and all the other read pairs are marked as duplicates and filtered out of downstream analyses. This helps avoid any systematic biases in the estimation of open chromatin due to PCR efficiencies for example. To mark duplicates each read pair is annotated with a start position, end position and its barcode. The start and end positions are adjusted for soft clipping. For computational efficiency reasons, we transform the barcode string into a 64-bit integer using a hash function. When a group of read pairs share the same (start, end, hashed barcode), one of them is labeled as unique and the rest are labeled duplicates. If the unique read passes the filters described in the next paragraph, this is the only read pair that is reported as a fragment in the fragment file. Note that versions of Cell Ranger ATAC <= 1.2 marked duplicates based on the start and end position alone. The previous algorithm was overly aggressive in marking duplicates as evidenced by the figure below. The estimated duplicate rate must be uniform throughout the genome and independent of the accessibility of the region. In the old method we observe that the duplicate rate actually increases as a function of accessibility, which is a signal that we were overly aggressive in marking duplicates.
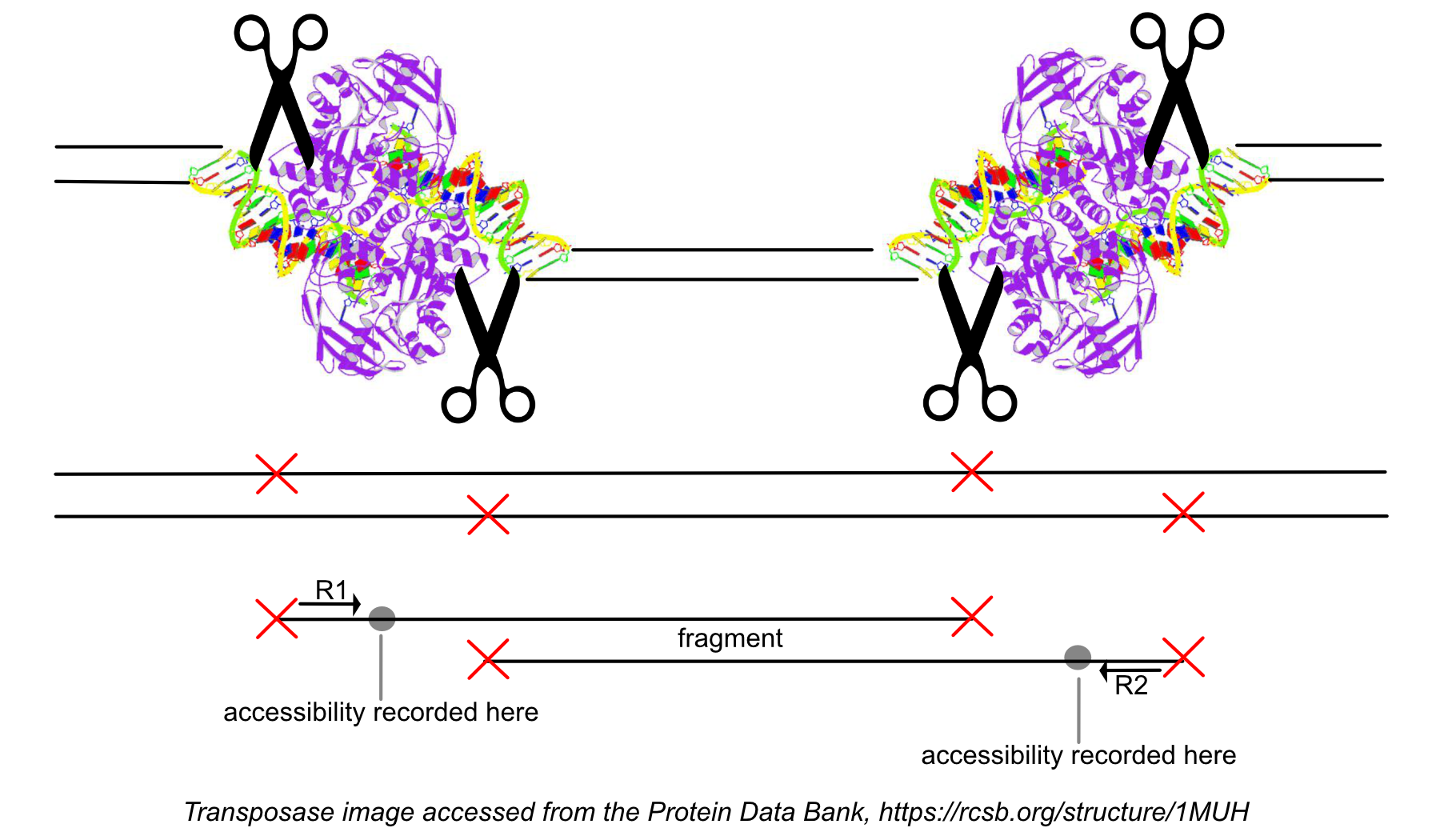
While processing the group of identically aligned read-pairs as described above, once the original fragment is marked, Cell Ranger ATAC determines if the fragment is mapped with MAPQ > 30 on both reads, is not mitochondrial, not chimerically mapped, and maps to a primary contig (a gene-containing contig). If the fragment passes these filters, Cell Ranger ATAC creates one entry in the fragments.tsv.gz file marking the start and end of the fragment after adjusting the 5' ends of the read-pair to account for transposition, during which the transposase occupies a region of DNA 9 base pairs long. Each entry is tab-separated and the file is position-sorted and then run through the SAMtools tabix command with default parameters.
The goal of the peak calling algorithm in the single-cell ATAC assay is to identify which distinct regions of the genome, known as peaks, are the key features of interest. This differs from single-cell gene expression assays, which use published gene annotations to define features. Called peaks in ATAC data are thus analogous to genes in gene expression data in the resulting feature-barcode matrix.
In the single-cell ATAC assay, the transposase enzyme cuts nuclear DNA at accessible regions, creating sequenceable fragments of DNA where each end identifies a transposase cut site. Because each sample may have cells with different patterns of chromatin accessibility, peaks must be called directly from ATAC data with each run of the pipeline. In order to accurately call peaks, the desired signal (open chromatin causing localized enrichment in cut sites) must be distinguished from background noise (random transposase activity throughout the genome). As both signal and noise can vary across different datasets and locally across the genome, the algorithm generates a global peak threshold followed by local refinements, as described below.
As the ends of each fragment are indicative of regions of open chromatin, Cell Ranger ATAC first analyzes the combined signal from these fragments, across all cells in the dataset, to determine regions of the genome enriched for accessibility to the transposase and thus of potential regulatory and functional significance. This method of identifying peaks uses reads pooled from all barcodes to maximize the signal from all mapped genomic fragments.
The fragment ends, corrected for the estimated binding position of the
transposase enzyme and de-duplicated, are identified in the position-sorted
fragments.tsv.gz file produced by Cell Ranger ATAC. Taking these data, the number
of transposition events at each position in the genome are counted. Because of
local variability in transposase binding affinity, this raw signal is smoothed
with a 401bp moving window sum to generate a smoothed signal profile, so that
the signal at each genomic position represents the total number of transposase
cut sites within the window around that position across all barcodes.
Genomic loci with higher counts are more likely to represent peaks than those with lower counts. To separate them, we count the number of genomic loci with each count and fit the underlying distribution to a mixture model of signal and noise components (figure below). The background is fit with a negative binomial distribution to capture stochastic noise and a geometric distribution to capture zero-inflation, similar to the Zero-Inflated Negative Binomial Algorithm (ZINBA). The peak signal is fit with a negative binomial distribution. The fitting is performed with an expectation-maximization iterative algorithm. The Cell Ranger ATAC 2.0 algorithm includes significant improvements to this fitting process to better initialize the fitting and avoid local maxima.
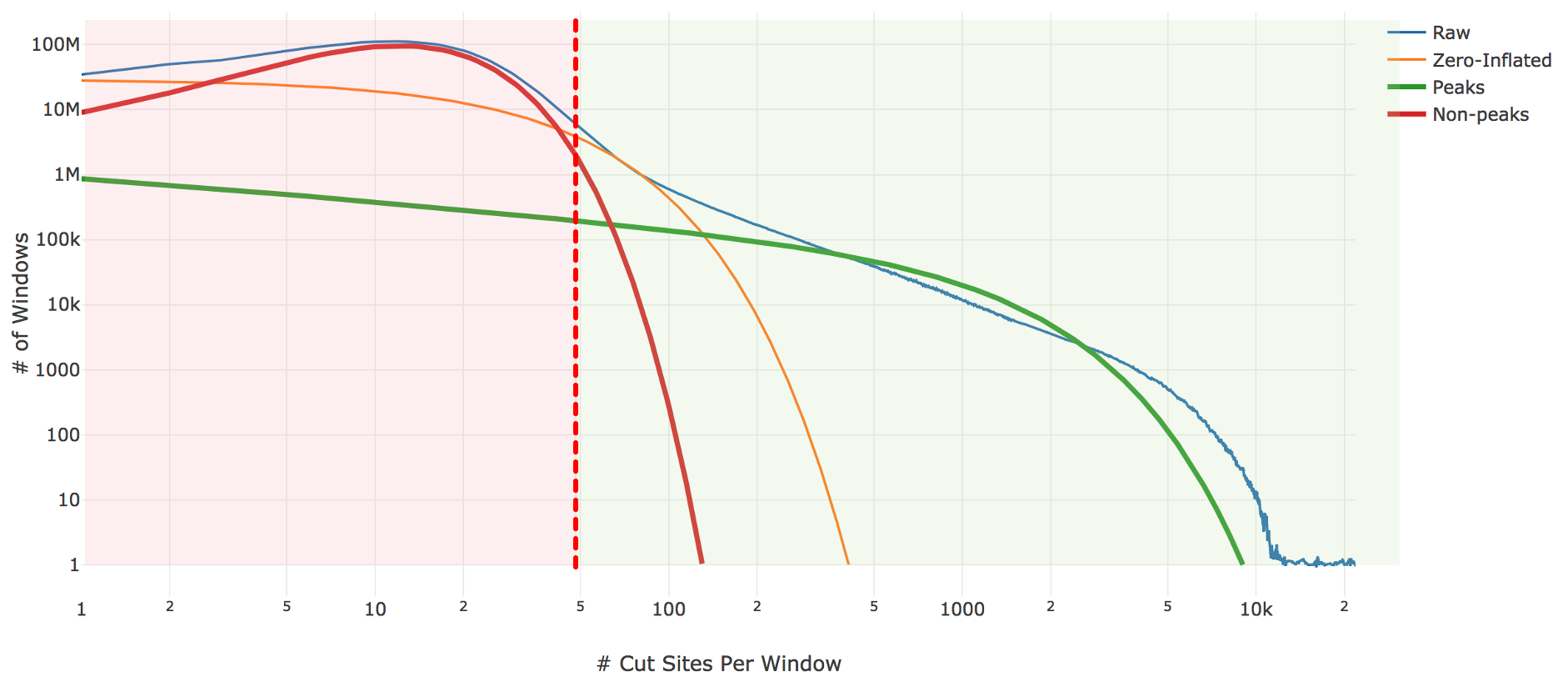
| Above: A diagram of the three-component fitting process for setting the initial global peak threshold. The x-axis shows (in logarithmic scale) the count of cut sites near a particular genomic locus, while the y-axis shows (again in logarithmic scale) the number of genomic windows with that cut-site count. The blue line is the observed data from our sample which the algorithm attempts to fit. The other three lines are the final fit: orange shows the geometric zero-inflated component, red the negative binomial non-peak background component, and green the negative binomial peak component. The sum of these three components closely approximates the empirical blue curve. The peak threshold (vertical red line) is set so at least 95% of the non-peak components are less than the threshold. |
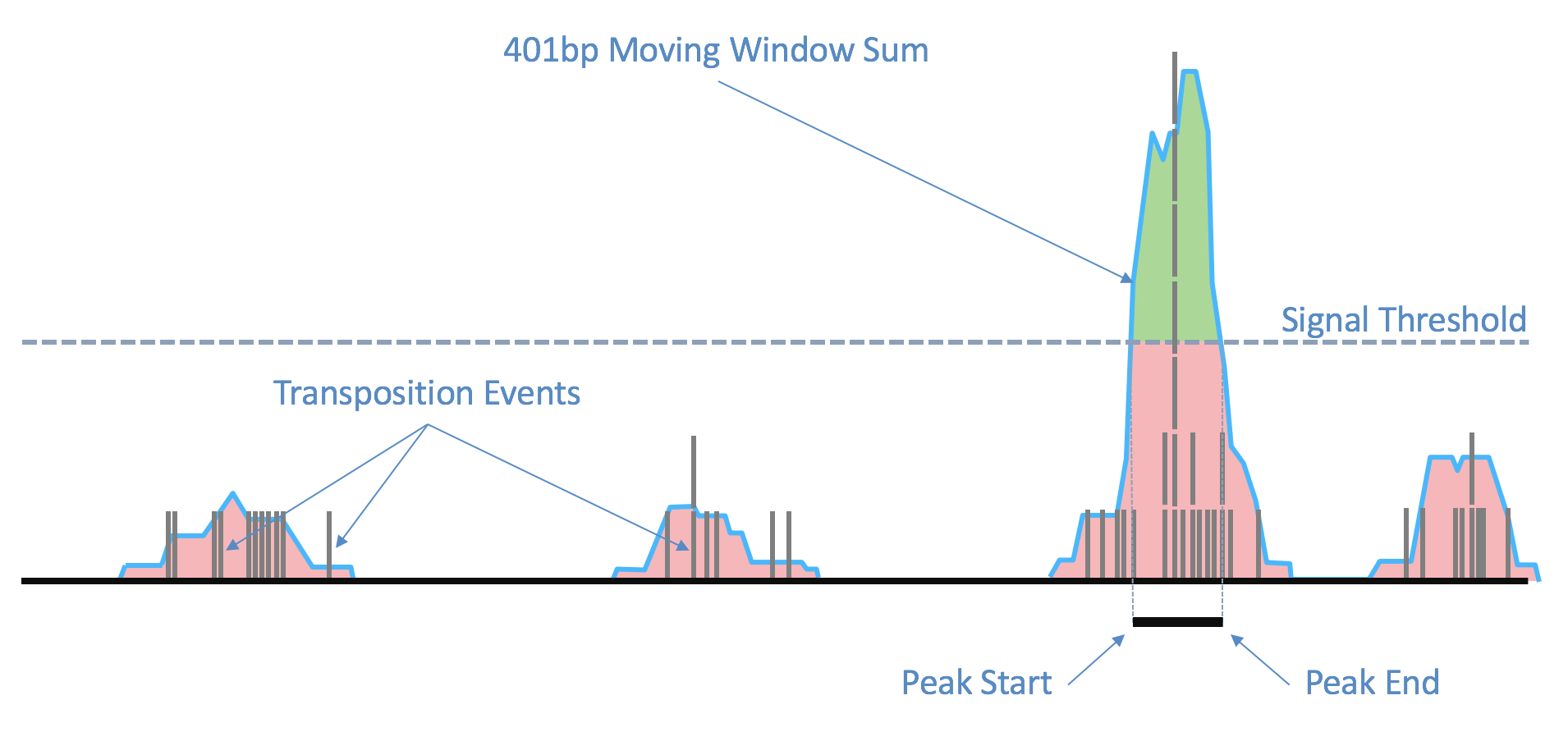
| Above: Raw transposition events are used to produce a local smoothed signal track with a 401bp moving window sum. After fitting and selecting a global peak threshold, contiguous regions with signal above the threshold (shown in green) are produced as candidate peak calls. |
In Cell Ranger ATAC v1.2 and earlier, the algorithm ended after global threshold generation, reporting as peaks all contiguous regions with smoothed signal above this global threshold. This sometimes produced peak calls that were too large, interfering with accurate identification of motifs and local regions of interest. Furthermore, the algorithm was susceptible to local differences in background noise across the genome. Therefore, in Cell Ranger ATAC 2.0 the algorithm includes further local refinement of candidate peak calls.
For each candidate region, the region is first padded by 5 Kb on either side to include local background context. The smoothed signal in the padded region is transformed with a 300bp Ricker wavelet transformation. Local maxima in the transformed signal are identified and putative peaks generated by extending the local maxima down to the total prominence of the maximum. Each putative peak is then filtered for local signal-to-noise ratio. The median signal inside the putative peak is used as the signal, while the noise is measured as the median signal in a fixed window outside of the peak, while also masking out any other putative peaks in the local region (figure below). Cell Ranger ATAC uses a Bayesian posterior probability estimate to exclude peaks that do not have a signal-to-noise ratio above 1.5 with at least 95% confidence. This Bayesian approach allows the use of a lower signal-to-noise threshold without overcalling peaks in very low depth regions.
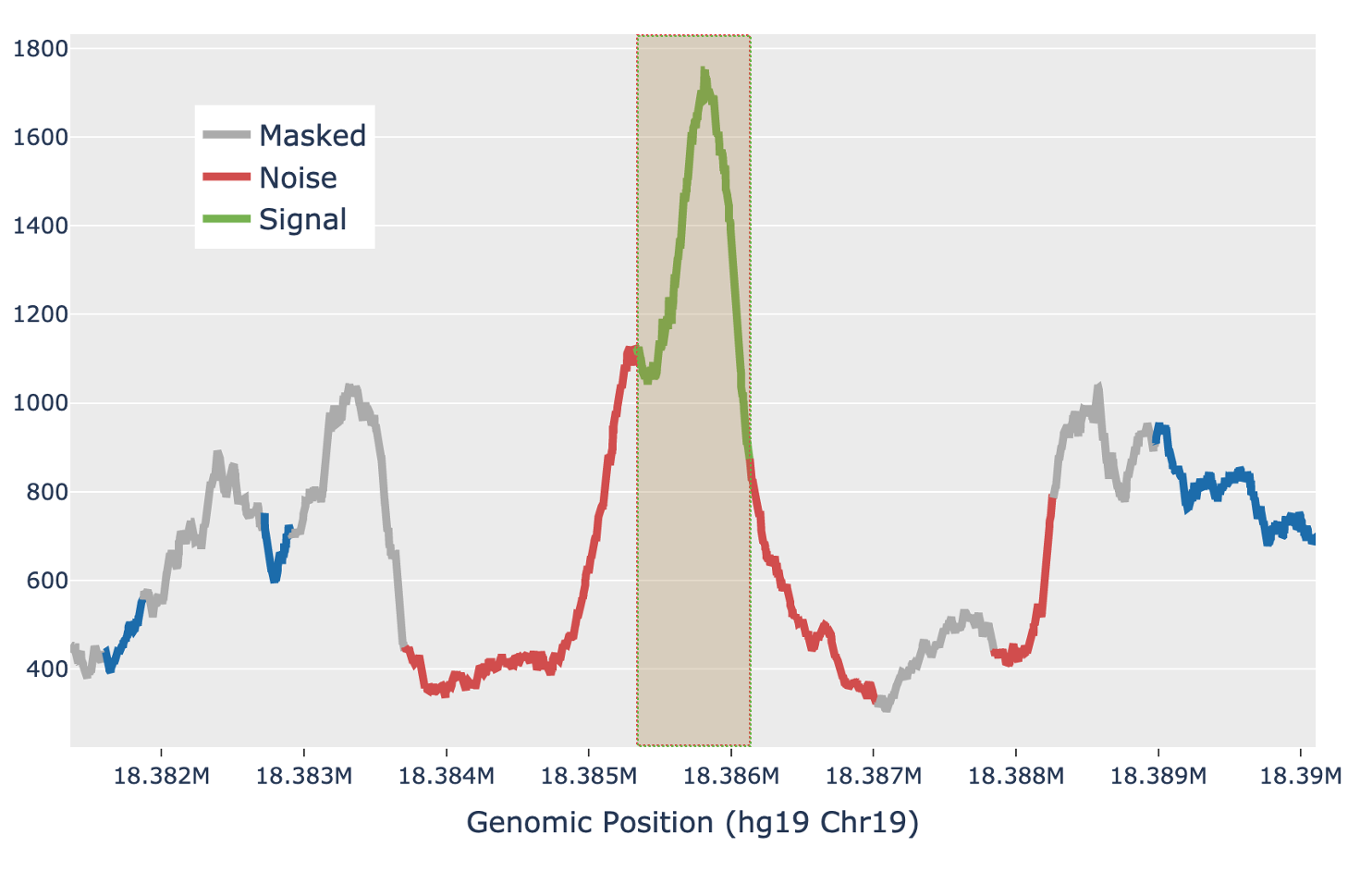
| Above: a diagram of how the local signal-to-noise estimate is performed for a single putative peak in a candidate region. The green section of the signal shows the putative peak under examination, with the peak signal measured as the median value across the green section. The grey sections are masked out, as they are other putative peaks and so are not used to estimate the local background. The red sections are used for local background estimates, with the peak background as the median value across all red sections. |
Finally, an extension step is performed on the filtered peaks. Cell Ranger ATAC examines all fragments inside a peak, each of which has two cut sites, one at each end. If there are a large number of fragments which have one cut site inside the peak and the other outside, the peak is padded to wholly contain more of those fragments.
This step associates a subset of barcodes observed in the library to the cells
loaded from the sample. Identification of these cell barcodes allows one to then
analyze the variation in data at single cell resolution. For each barcode, we
have the record of mapped high-quality fragments that passed all filters (the
fragments.tsv file). Having determined peaks prior to this, we use the number of
fragments that overlap any peak regions, for each barcode, to separate the
signal from noise. This works better in practice as compared to naively using
the number of fragments per barcode. The cell calling is done in two steps.
First we identify barcodes that have fraction of fragments overlapping called
peaks lower than the fraction of genome in peaks (for the sake of this
calculation only, peaks are padded by 2000 bp on both sides to account for
fragment length). We have found that these barcodes typically have their cut
sites randomly distributed over the genome, are not targeted to be enriched near
functional regions, and do not exhibit the expected ATAC-seq "peaky" signal.
Therefore we mask these low targeting barcodes out of the total set of
barcodes observed for the library prior to cell calling.
The 10x Chromium system has a low rate of gel bead multiplets (predominantly doublets) where a cell shares more than one barcoded gel bead. These cells then manifest as multiple barcodes of the same cell type in the dataset. The presence of these few extra barcodes doesn't affect secondary analysis such as clustering or differential analysis, although it can potentially inflate abundance measurements of very rare cell types. We identify a minor-major pair of barcodes (B1, B2) that are part of a putative gel bead doublet by observing if the pair of barcodes shares more genomically adjoining "linked" fragments (fragments sharing a transposition event) with each other (B1-B2) as opposed to themselves (B1-B1 or B2-B2). The minor barcode is identified as the one with fewer fragments and discarded from the set of total barcodes used in cell calling.
Single Cell ATAC data also has another source capable of generating extra cells of similar kind. This phenomenon is known as barcode multiplets, which occurs when a cell associated gel bead is not monoclonal and has the presence of more than one barcode. The barcodes associated with such multiplets are identified as ones sharing significant number of linked fragments with each other as well as having a common suffix or a prefix nucleotide sequence. Again, we mask out the "minor" barcode participating in these multiplets while retaining the major barcode as the sole representative of the associated cell.
We then perform cell calling on the remainder barcodes. We subtract a depth-dependent fixed count from all barcode counts to model whitelist contamination. This fixed count is the estimated number of fragments per barcode that originated from a different GEM, assuming a contamination rate of 0.02. Then we fit a mixture model of two negative binomial distributions to capture the signal and noise. Setting an odds ratio of 100000 (which appeared to work best with internal testing), we separate the barcodes that correspond to real cells from the non-cell barcodes.
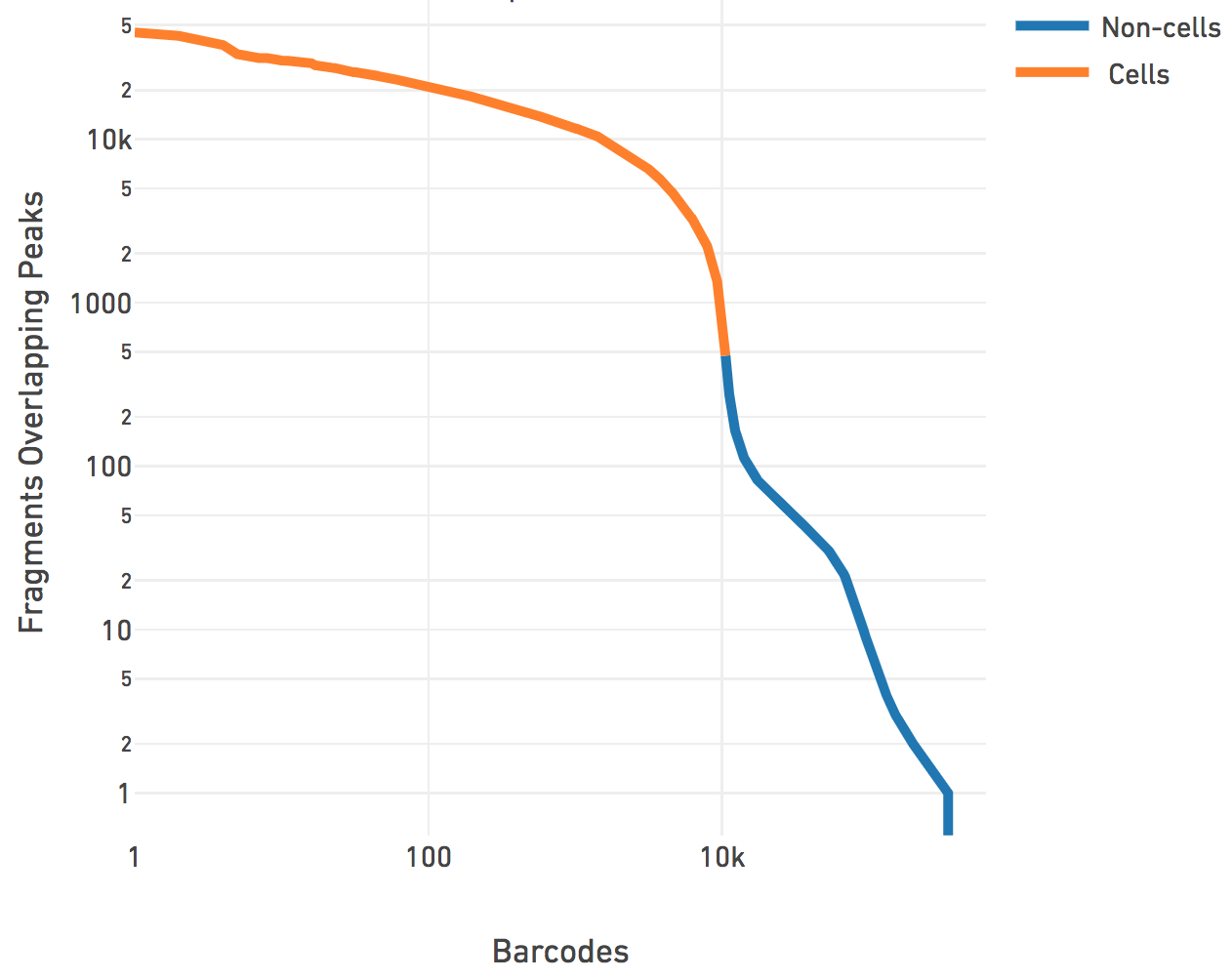
The cell calling is limited to produce < 20k cells per species in the reference
as the assay is currently designed to support 500-10k cells. If
--force-cells=N is provided as a parameter to Cell Ranger ATAC, we
ensure that the top N barcodes are reported as cells for each species, as per
the barcode rank plot above. N > 20k will not be accepted by the
pipeline. If --force-cells is not provided, in the case of mixed
species sample, we do a second iteration where we mask out the non-cell barcodes
and fit the same mixture model to the two species distributions present in the
cell barcodes and refine the division of cell barcodes associated with each
species. In general, the --force-cells value to be used should
correspond to a point below the "knee" as seen on the barcode rank plot above.
Similar to our analysis pipelines for the Single Cell Gene Expression Solution and the Single Cell Immune Profiling Solution, we produce a count matrix consisting of the counts of fragment ends (or cut sites) within each peak region for each barcode. This is the raw peak-barcode matrix and it captures the enrichment of open chromatin per barcode. The matrix is then filtered to consist of only cell barcodes, which is then used in subsequent analysis such as dimensionality reduction, clustering and visualization.
Biological discovery is often aided by visualization tools that allow one to
group and compare a population of cells with another. In order to enable
discovery, Cell Ranger ATAC performs clustering and t-SNE projection. As the data is
sparse at single cell resolution, we first perform dimensionality reduction to
cast it into a lower dimensional space, which also has the benefit of
de-noising. Cell Ranger ATAC supports dimensionality reduction via Principal
Component Analysis (PCA), Latent Semantic Analysis (LSA), or Probabilistic
Latent Semantic Analysis (PLSA). The adopted default method is LSA, but users
can specify which method to use by providing the dimensionality reduction
parameter (--dim-reduce=<Method>) to Cell Ranger ATAC. Each of these
methods acts on the filtered peak-barcode matrix consisting of cut site counts
for cell barcodes in called peaks. Each method has an associated data
normalization technique used prior to dimensionality reduction and a collection
of clustering methods that accept the data after dimensionality reduction. We
also provide an optimized implementation of the Barnes Hut
TSNE algorithm (which is the same as the one
in our analysis pipeline for the Single Cell Gene Expression Solution). The
number of dimensions is fixed to 15 as it was found to sufficiently separate
clusters visually and in a biologically meaningful way when tested on peripheral
blood mononuclear cells (PBMCs).
For PCA, we first normalize the data to median cut site counts per barcode and log-transform it. We use a fast, scalable and memory efficient implementation of IRLBA (Augmented, Implicitly Restarted Lanczos Bidiagonalization Algorithm) that allows in-place centering and feature scaling and produces the transformed matrix along with the principal components (PC) and singular values encoding the variance explained by each PC. Specific to PCA, we provide k-means clustering that produces 2 to 10 clusters for visualization and analysis. We also provide a k-nearest neighbors graph-based clustering method via community detection using louvain modularity optimization algorithm. The transformed matrix is operated on by the t-SNE algorithm with default parameters and provides 2-D coordinates for each barcode for visualization. Users experienced with our Single Cell Gene Expression Solution may recognize that the analysis performed using PCA is akin to running Cell Ranger (cellranger count).
Inspired by the large body of work in the field of information retrieval, we normalize the data via the inverse-document frequency (idf) transform where each peak count is scaled by the log of the ratio of the number of barcodes in the matrix and the number of barcodes where the peak has a non-zero count. This provides greater weight to counts in peaks that occur in fewer barcodes. Singular value decomposition (SVD) is performed on this normalized matrix using IRLBA without scaling or centering, to produce the transformed matrix in lower dimensional space, as well as the components and the singular values signifying the importance of each component. Prior to clustering, we perform normalization to depth by scaling each barcode data point to unit L2-norm in the lower dimensional space. We found that the combination of these normalization techniques obviates the need to remove the first component. Specific to LSA, we provide spherical k-means clustering that produces 2 to 10 clusters for downstream analysis. Spherical k-means was found to perform better than plain k-means, by identifying clusters via k-means on L2-normalized data that lives on the spherical manifold. Similar to PCA, we also provide a graph-based clustering and visualization via t-SNE. However, similar to spherical k-means clustering, we normalize the data to unit norm before performing graph-based clustering and t-SNE projection.
PLSA is a special type of non-negative matrix factorization, with roots in Natural Language Processing. In PLSA we minimize, via an Expectation-Maximization algorithm, the KL-divergence between the empirically determined probability of observing a peak in a barcode and the lower rank approximation to it. We do not normalize the data prior to dimensionality reduction via PLSA. Similar to LSA and PCA, we produce a transformed matrix, component vectors and a set of values explaining the importance of each component. PLSA offers natural interpretation of the components and the transformed matrix. Each component could be interpreted as a hidden topic and the transformed matrix is simply the probability of observing a barcode from a given topic, i.e. Prob(barcode|topic). The component vectors are the probability of observing a peak from a given topic (Prob(peak|topic)) and the counterpart to singular values of LSA/PCA are simply the probability of each topic (Prob(topic)) observed in the data. Similar to LSA, we normalize the transformed matrix to unit L2-norm and perform spherical k-means clustering to produce 2 to 10 clusters, as well as graph-based clustering and visualization via t-SNE. While PLSA offers great advantages in interpretability of the lower dimensional space, it is appreciably slower than both PCA and LSA and does not scale well beyond 20 components on large datasets. To ameliorate this to some extent, the in-house implementation of PLSA is multithreaded (4 threads on compute cluster) and written and compiled in C++. To ensure a reasonable run time, the algorithm is capped at 3000 iterations if it doesn't converge first.
Below is the summary of dimensionality reduction techniques and associated clustering and visualization approaches provided in the pipeline.
| Dimensionality Reduction | Clustering | Visualization |
|---|---|---|
| PCA | K-means, graph-clustering | TSNE |
| LSA | Spherical k-means, graph-clustering | TSNE |
| PLSA | Spherical k-means, graph-clustering | TSNE |
| Note that in version 1.0 of the Cell Ranger ATAC pipelines, we provided k-medoids clustering. Spherical k-means was found to be an effective replacement for k-medoids for both LSA and PLSA, with a significant performance gain that makes it suitable to cluster large scale datasets you can expect from aggregation runs. |
As peaks are regions enriched for open chromatin, and thus have potential for regulatory function, observing the location of peaks with respect to genes can be insightful. We use bedtools closest -D=b to associate each peak with genes based on closest transcription start sites (packaged within the reference) such that the peak is within 1000 bases upstream or 100 bases downstream of the TSS. Additionally, we also associate genes to putative distal peaks that are much further from the TSS, and are less than 100kb upstream or downstream from the ends of the transcript. This association is adopted by our companion visualization software (Loupe Browser) and used to construct and visualize derived features such as promoter-sums that pool together counts from peaks associated with a gene. Peaks are enriched for transcription factor (TF) binding motifs and the presence of certain motifs can be indicative of transcription factor activity. To identify these motifs, we first calculate the GC% distribution of peaks and then bin the peaks into equal quantile ranges in the GC content distribution. We use the MOODS Python library packaged inside Cell Ranger ATAC to scan each peak for matches to motif position-weight-matrices (PWMs) for transcription factors from the JASPAR database built directly into the reference package. We set a p-value threshold of 1E-7 and background nucleotide frequencies to be the observed nucleotide frequencies within the peak regions in each GC bucket. The list of motif-peak matches is unified across these buckets, thus avoiding GC bias in scanning.
As transcription factors (TF) tend to bind at sites containing their cognate motifs, grouping of accessibility measurements at peaks with common motifs produces a useful enrichment analysis of TFs across single cells. We construct an integer count for each TF for each cell barcode in the following manner: we consider all peaks matched to a given TF, as discovered in the TF motif detection. Then for every barcode, we pool together the cut-site counts across these matched peaks in the filtered peak-barcode matrix. This calculates the total cut-sites in a cell barcode for peaks that share the TF motif. We calculate the proportion of cut-sites for a TF within a barcode out of the total cut-sites for that barcode, which normalizes it to depth. We detect enrichment for a TF by z-scoring the distribution over barcodes of these proportion values for given TF. To make it robust to outliers, we use the modified z-score calculated using the median and the scaled median absolute deviation from the median (MAD), instead of the mean and standard deviation. These z-scored values are visible when you load a dataset in Loupe and accompanies the differential analysis built into Loupe Browser.
| We do not produce the tf-barcode matrix for multi-species experiments or if the motifs.pfm file is missing from the reference package (for example in custom references). We cannot perform TF motif enrichment analysis in these cases. |
In order to identify transcription factor motifs whose accessibility is specific to each cluster, Cell Ranger ATAC tests, for each motif and each cluster, whether the in-cluster mean differs from the out-of-cluster mean. Users familiar with our Single Cell Gene Expression Solution may recognize that this is identical to what Cell Ranger does for identifying differential gene expression. In order to find differentially accessible motifs between groups of cells, Cell Ranger ATAC uses a Negative Binomial (NB2) generalized linear model to discover the cluster specific means and their standard deviations, and then employs a Wald test for inference. For each cluster, the algorithm is run on that cluster versus all other cells, yielding a list of TF motifs that are differentially expressed in that cluster relative to the rest of the sample. Using a GLM framework allows us to model the sequencing depth per cell and GC content in peaks per cell directly as covariates. This allows us to normalize to them naturally as part of model estimation and inference procedure. We also perform differential enrichment analysis for accessibility in peaks using a Poisson generalized linear model, much the same way as we do for TF motifs. In this case, we model only the per cell depth as a covariate.
| We cannot perform differential analysis for transcription factor motifs in the cases where the motifs.pfm file is missing from the reference package, such as in custom references built without the motif file or in multi-species experiments. We will always perform differential analysis on accessibility in peaks for single species references. |
In the aggr pipeline, you can provide a list of libraries to aggregate. The aggregation pipeline performs the task of merging the fragments from each listed library into one aggregated file, based on the normalization mode specified at runtime. By default, all the fragments are retained and merged together. If the normalization mode is set to "depth", then each library is downsampled to have the same sensitivity (defined as per cell median number of fragments). Once the fragments are merged together, they are sorted by position and tabixed for downstream use such as dimensionality reduction, clustering, visualization and differential analysis.