Cell Ranger ATAC1.0, printed on 03/29/2025
Cell Ranger ATAC is a set of analysis pipelines that process Chromium Single Cell ATAC data. Cell Ranger ATAC includes two pipelines relevant to single cell chromatin accessibility experiments:
cellranger-atac mkfastq demultiplexes raw base call (BCL) files generated by Illumina® sequencers into FASTQ files. It is a wrapper around bcl2fastq from Illumina®, with additional useful features that are specific to 10x Genomics libraries and a simplified sample sheet format.
cellranger-atac count takes FASTQ files from cellranger-atac mkfastq and performs ATAC analysis, including:
Output is delivered in standard BAM, MEX, CSV, TSV, HDF5 and HTML formats that are augmented with cellular information.
The count pipeline can take input from multiple sequencing runs on the same library.
Throughout the documentation, you will see references to samples, libraries and sequencing runs (see the Glossary of Terms for detailed definitions). The relationship between these terms can be complex as there are multiple ways samples can be prepared for the pipeline.
Cell Ranger ATAC 1.0 supports libraries generated by the Chromium Single Cell ATAC v1 reagent kits.
10x Genomics recommends using the pipeline analysis programs in order, staring with cellranger-atac mkfastq for demultiplexing the raw base call (BCL) files for each flowcell directory, and continuing with cellranger-atac count for single library analysis. If compatible FASTQ files are available from another source, a user can skip cellranger-atac mkfastq and use those FASTQ files as direct input to cellranger-atac count. Compatible FASTQ files can be found in reputable public datasets, or can be built by using bcl2fastq directly. See the Specifying Input FASTQs page for more details
The subsequent steps vary depending on how many samples, libraries, and flowcells you have. They are described here in order of increasing complexity:
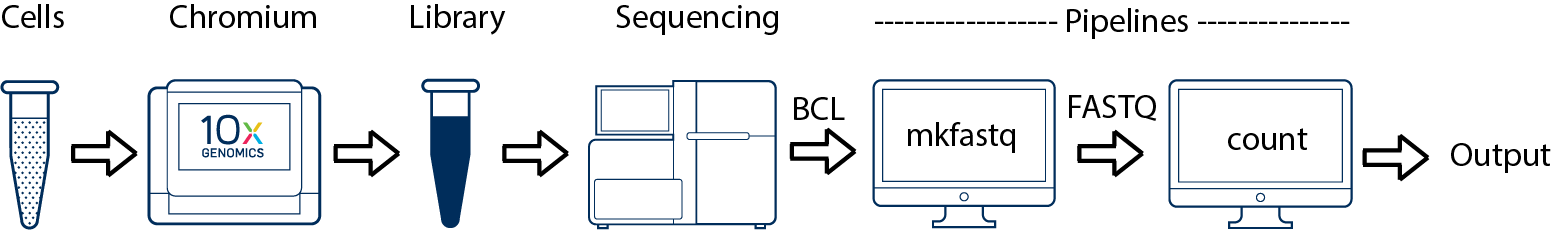
This is the most basic case. You have a single biological sample, which was prepared into a single library, and then sequenced on a single flowcell. Assuming the FASTQs have been generated with cellranger-atac mkfastq, you just need to run cellranger-atac count as described in Single-Library Analysis.
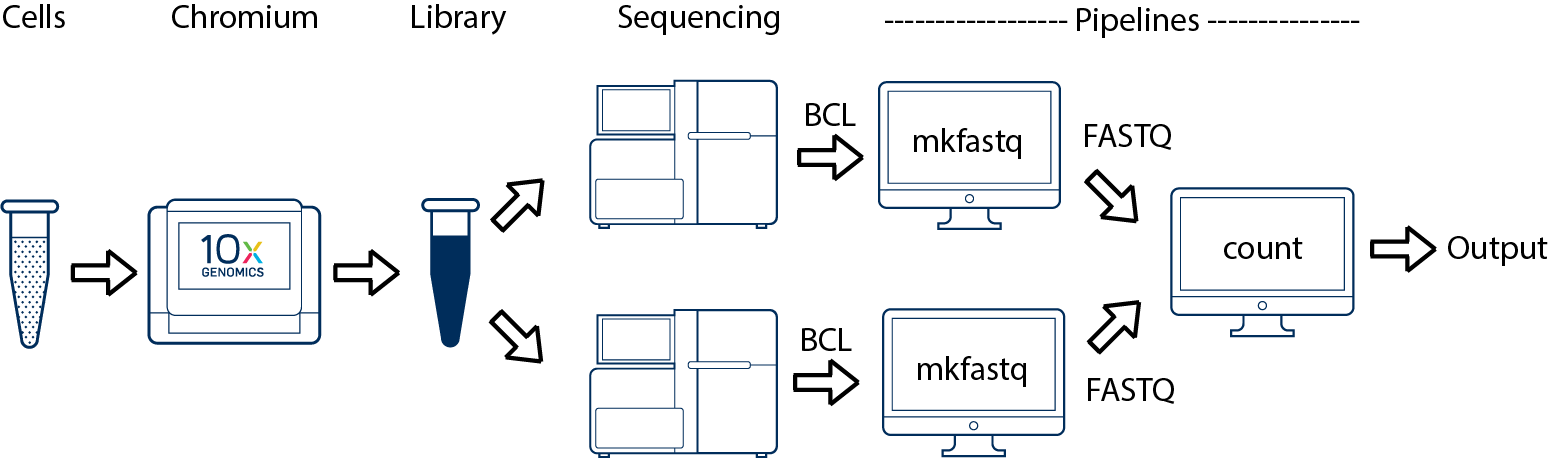
If you have a library which was sequenced across multiple flowcells (e.g. to increase sequencing saturation), you can pool the reads from both sequencing runs. Follow the steps in Specifying Input Fastqs to combine them in a single cellranger-atac count run.