Cell Ranger4.0, printed on 04/06/2025
Targeted Gene Expression analysis is available in Cell Ranger 4.0 and is invoked by specifying the --target-panel option when running the cellranger count command.
Cell Ranger 4.0 introduces the new targeted-compare pipeline for direct comparative analysis of matched parent Whole Transcriptome Amplification (WTA) and Targeted Gene Expression datasets.
Cell Ranger 4.0 includes the new targeted-depth subcommand to estimate sequencing depths appropriate for Targeted Gene Expression experiments based on input WTA results and an associated target panel file.
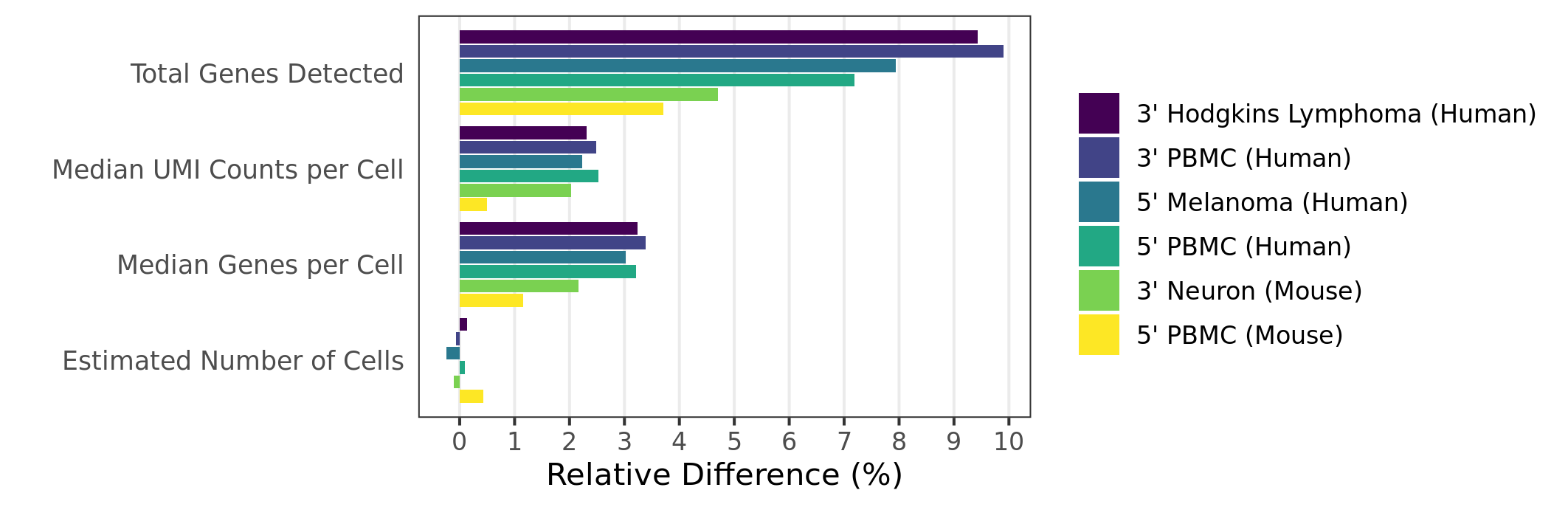
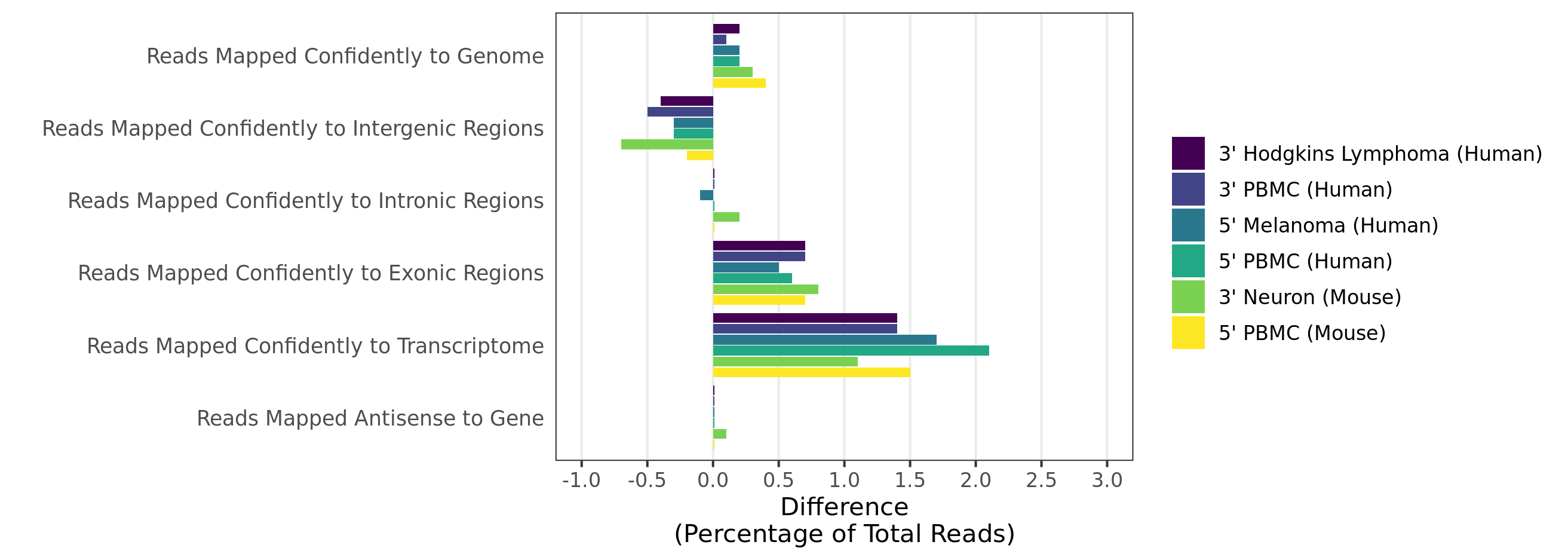
Recommended reference packages for human and mouse have been updated from version 3.0.0 to 2020-A:
chr1 and chrM) rather than the Ensembl convention (1 and MT).Mapping rates and gene/UMI sensitivity are increased due to more comprehensive annotations and improved manual curation of genes:
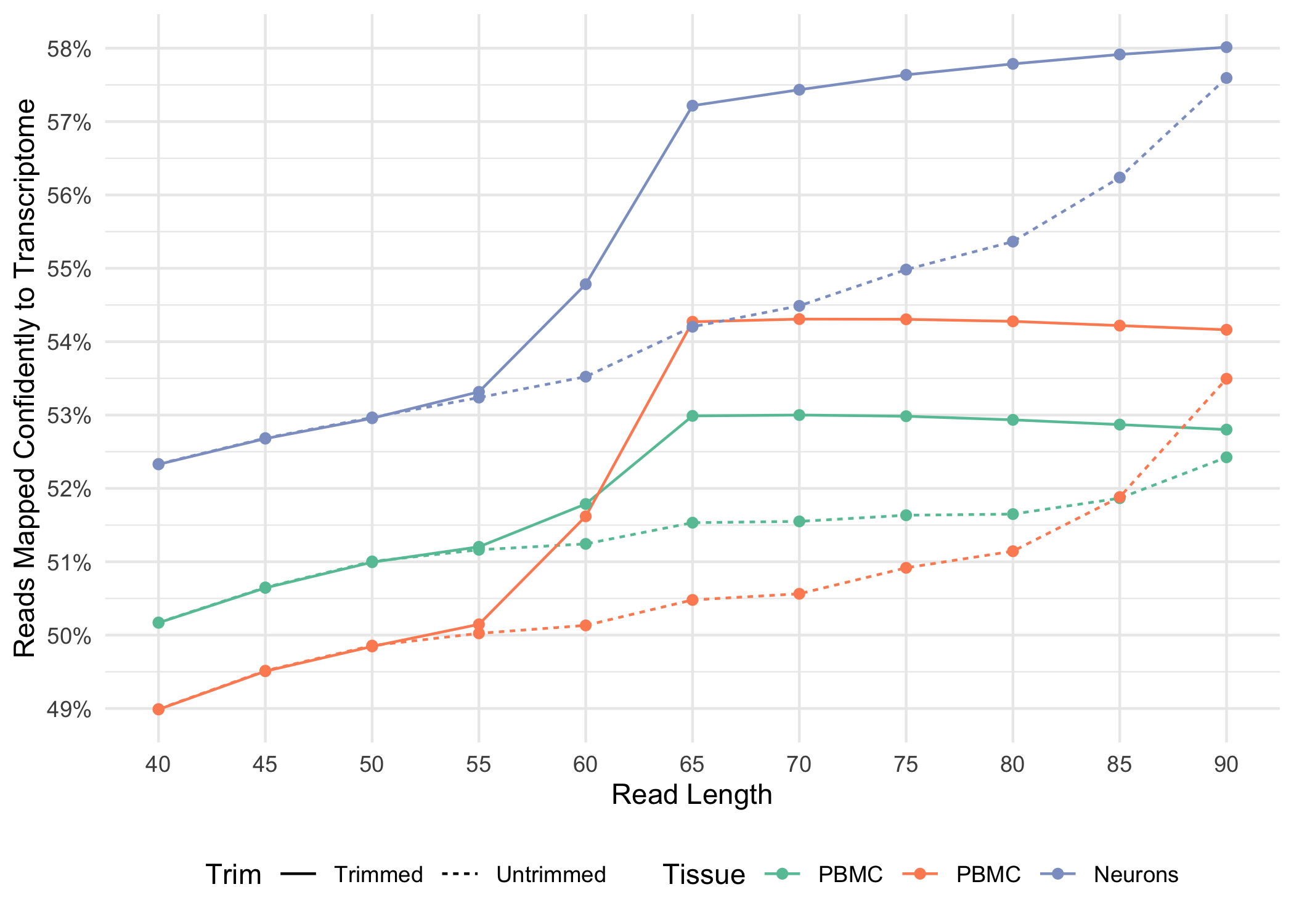
When analyzing 3’ Gene Expression data, Cell Ranger 4.0 trims the template switch oligo (TSO) sequence from the 5’ end of Read-2 and the poly-A sequence from the 3’ end before aligning reads to the reference transcriptome. This behavior is different from Cell Ranger 3.1, which does not perform any trimming.
A full length cDNA molecule is normally flanked by the 30-bp TSO sequence, AAGCAGTGGTATCAACGCAGAGTACATGGG, at the 5' end and the poly-A sequence at the 3' end. Some fraction of sequencing reads are expected to contain either or both of these sequences, depending on the fragment size distribution of the library. Reads derived from short RNA molecules are more likely to contain either or both TSO and poly-A sequence than longer RNA molecules.
Trimming results in better alignment, with the fraction of reads mapped to a gene increasing by up to 1.5%, because the presence of non-template sequence in the form of either TSO or poly-A confounds read mapping. Trimming improves the sensitivity of the assay as well as the computational efficiency of the pipeline. Tags ts:i and pa:i in the output BAM files indicate the number of TSO nucleotides trimmed from the 5' end of Read-2 and the number of poly-A nucleotides trimmed from the 3' end. The trimmed bases are present in the sequence of the BAM record and are soft clipped in the CIGAR string.
Below, we illustrate how the fraction of reads mapped confidently to the transcriptome varies for both trimmed and untrimmed alignment as a function of read-length for a variety of sample types
.
Cell Ranger 4.0 adds support for an “un-tethered” Feature Barcode pattern, (BC) without an anchor, specified in the Feature Reference CSV. This option allows the user to specify the sequence of the Feature Barcode without specifying a particular location on the read where the sequence is expected to be found.
cellranger reanalyze now outputs the count matrix used in the analysis, so as to reflect any subsetting of barcodes used.
Bug fixes for GTF files output by mkref. These changes do not affect the pipeline results.
tag "value1"; tag "value2";) are handled correctly. Previously, only the last such attribute was kept.exon_number 1;) are kept. Previously, they were removed.Bug fixes for the BAM file
0x400) is set correctly in the secondary alignments (flag 0x100) of PCR duplicate reads and low-support UMI reads (xf:i:2)xf:i:2) have the corrected barcode in UB:Z. Previously, it contained the raw barcode.BAM file changes
li:i tag. The RG:Z tag contains this information.BC:Z and QT:Z tags.Cell Ranger 4.0 now relies on Orbit to perform transcriptome alignment, which leverages a modified STAR v2.7.2a. These modifications provide compatibility with “versionGenome 20201” references, such as those generated by STAR v2.5.1b. In Cell Ranger 4.0 we still provide and use STAR v2.5.1b for other purposes such as cellranger mkref. In our testing we did not note any differences in transcriptome alignments between the STAR shipped in Cell Ranger 3.1 (STAR v2.5.1b), STAR v2.7.2a, or Orbit.
SC3Pv1 or SC5P-R1 respectively) using the --chemistry argument.--chain is added back in 4.0 for rare cases when the automatic chain detection fails.airr_rearrangement.tsv is added, which contains annotated contigs of VDJ rearrangements in the AIRR TSV format.