Cell Ranger ARC1.0, printed on 04/06/2025
The computational pipeline cellranger-arc count for Single Cell Multiome ATAC + Gene Expression involves the following analysis steps:
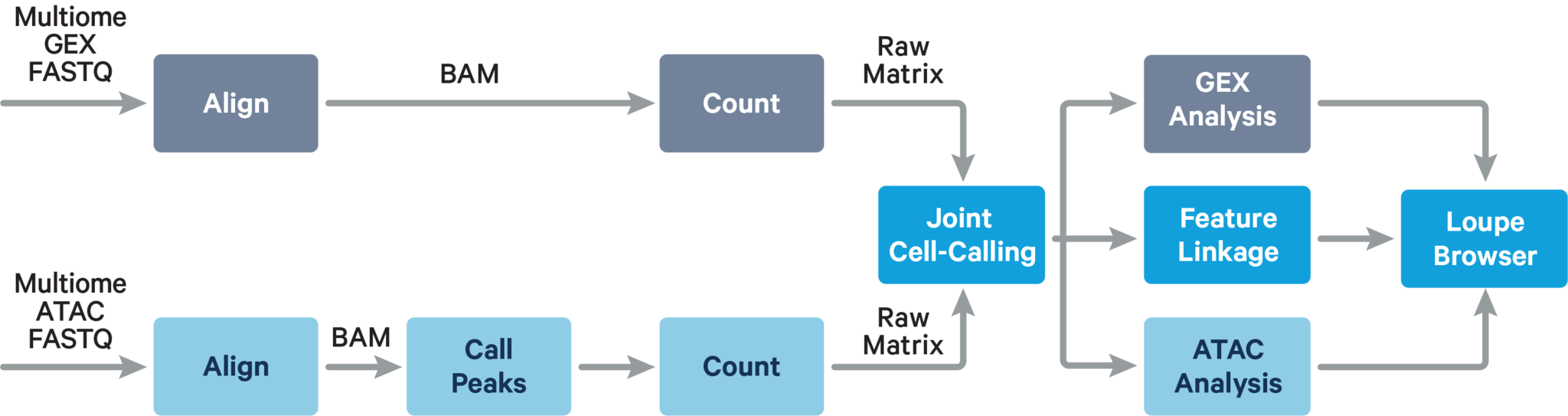
The main components of cellranger-arc count include:
For ATAC reads, Cell Ranger ARC performs reference-based analysis and requires adapter and primer oligo sequence to be trimmed off before mapping confidently. In the current chemistry, the 3' end of a read (the end of the read sequence) may contain the reverse complement of the primer sequence, if the read length is greater than the length of the genomic fragment. We use the cutadapt tool to identify the reverse complement of the primer sequence at the end of each read, and trim it from the read prior to alignment.
Then, the trimmed read-pairs are aligned to a specified reference using BWA-MEM with default parameters. Please note that BWA-MEM will not align reads less than 25 bp. These unaligned reads are included in the BAM output and are flagged as unmapped.
This step is performed to fix the occasional sequencing error in barcodes so that fragments get associated with the original barcodes, thus improving data quality. The 16 bp barcode sequence is obtained from the "I2" index read. Each barcode sequence is checked against a 'whitelist' of correct barcode sequences and the frequency of each whitelist barcode is counted. We attempt to correct barcodes that aren't on the whitelist, by finding all whitelisted barcodes that are within 2 differences (Hamming distance <= 2) of the observed sequence, and scoring them based on the abundance of that barcode in the read data and quality value of the incorrect bases. An observed barcode not present in the whitelist is corrected to a whitelist barcode if it has > 90% probability of being the real barcode based on this model.
A barcoded fragment may get sequenced multiple times due to PCR amplification. Duplicates are marked to identify the original fragments that constitute the library and contribute to its complexity. Duplicate reads are found by identifying groups of read-pairs, across all barcodes, where the 5' ends of both R1 and R2 have identical mapping positions on the reference, correcting for soft-clipping. These groups of read pairs arise from the same original molecule. Among these read-pairs, the most common barcode sequence is identified. One of the read-pairs with that barcode sequence is labelled the 'original' and the other read pairs in the group are marked as duplicates of that fragment in the BAM file. If the read pair passes the filters described in the next paragraph, it is the only read pair that is reported as a fragment in the fragment file and is marked with the most common barcode sequence.
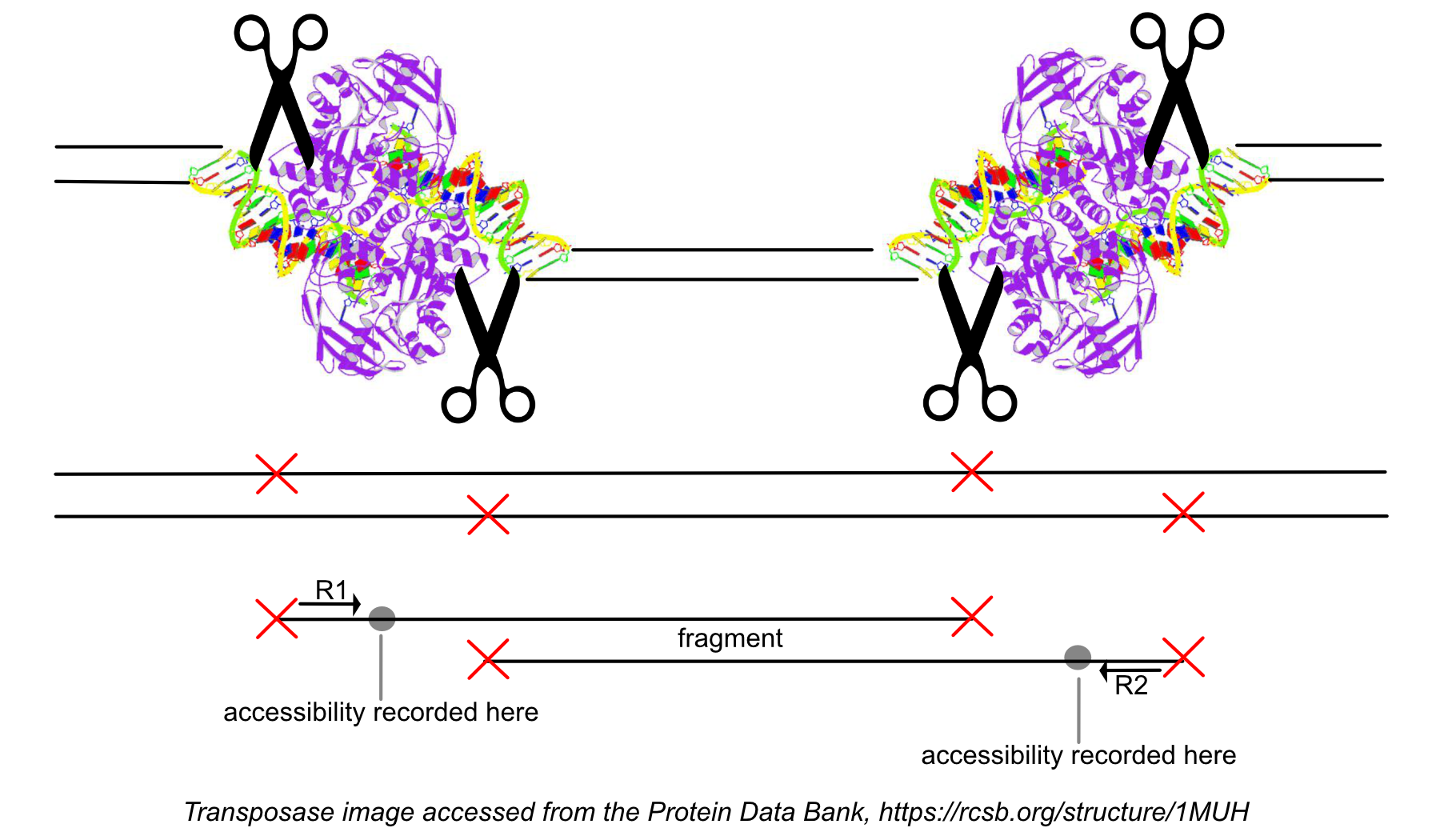
After the 'original' fragment is marked while processing the group of identically aligned read pairs described above, we determine if the fragment is mapped with MAPQ >30 on both reads, is not mitochondrial, and not chimerically mapped. If the fragment passes these filters, we create one entry in the fragments.tsv.gz file marking the start and end of the fragment after adjusting the 5' ends of the read pair to account for transposition, during which the transposase occupies a 9 bp DNA region (see figure). With this entry, we associate the most common barcode observed for the group of read pairs and the number of times this fragment is observed in the library (size of the group). Note that as a consequence of this approach, each unique interval on the genome can be associated with only one barcode. Each entry is tab-separated and the file is position-sorted and then run through the SAMtools tabix command with default parameters.
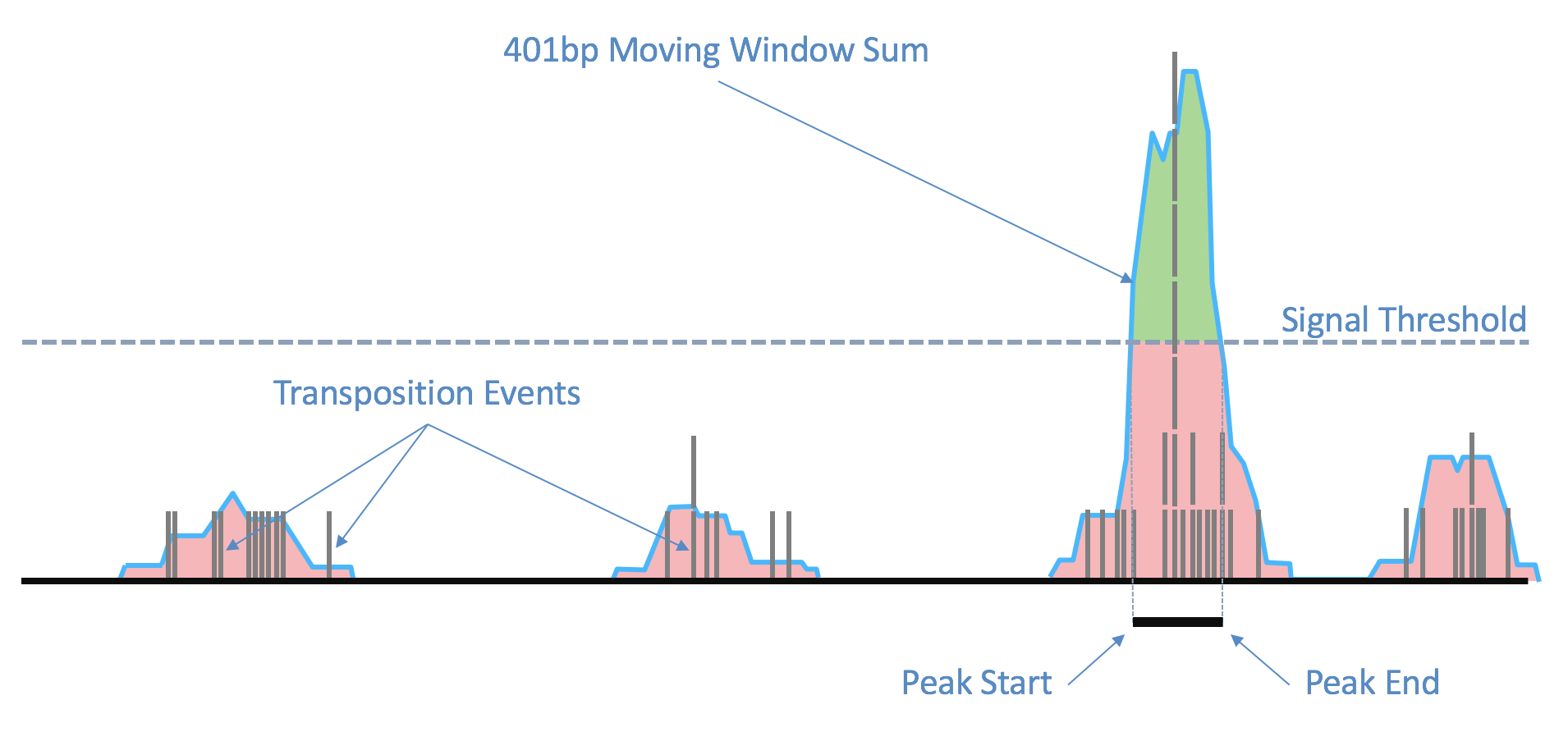
As the ends of each fragment are indicative of regions of open chromatin, we analyze the combined signal from these fragments to determine regions of the genome enriched for open chromatin and thus, putatively of regulatory and functional significance. Using the sites as determined by the ends of the fragments in the position-sorted fragments.tsv.gz file, we count the number of transposition events at each base-pair along the genome. We generate a smoothed profile of these events with a 401bp moving window sum around each base-pair and fit a ZINBA-like mixture model consisting of geometric distribution to model zero-inflated count, negative binomial distribution to model noise and another geometric distribution to model the signal. The fitting is performed in a way to ensure a fixed ordering of the means of mixture components: first the negative binomial noise mean, followed by geometric zero-inflation mean and finally by the geometric signal distribution mean. We set a signal threshold based on an odds-ratio of 1/5 that determines, at base pair resolution, whether a region is a peak signal (enriched for open chromatin) or noise. Consequently, not all cut sites are within a peak region. Peaks within 500bp of each other are merged to produce a position-sorted BED file of peaks. The window size, the odds-ratio and the distance to merge peaks were selected to maximize the area under the receiver-operator curve when the peaks are compared to a high-quality list of DNase hypersensitive sites for GM12878 from ENCODE, and also to produce satisfactory clustering metrics as well as cell type identification in a set of peripheral blood mononuclear cells (PBMC) libraries. This method of identifying peaks is independent of barcodes and their cellular (or non-cell) identity, which allows us to include the signal from all real genomic fragments as determined by mapping. A theoretical disadvantage of such a method would be not being able to identify rare peaks appearing only in very rare populations.
Once the peak locations have been determined we construct the peak-barcode matrix using peaks as the rows and barcodes as the columns. Each entry in this matrix is the number of cut-sites that are contained within the corresponding peak associated with the corresponding barcode.
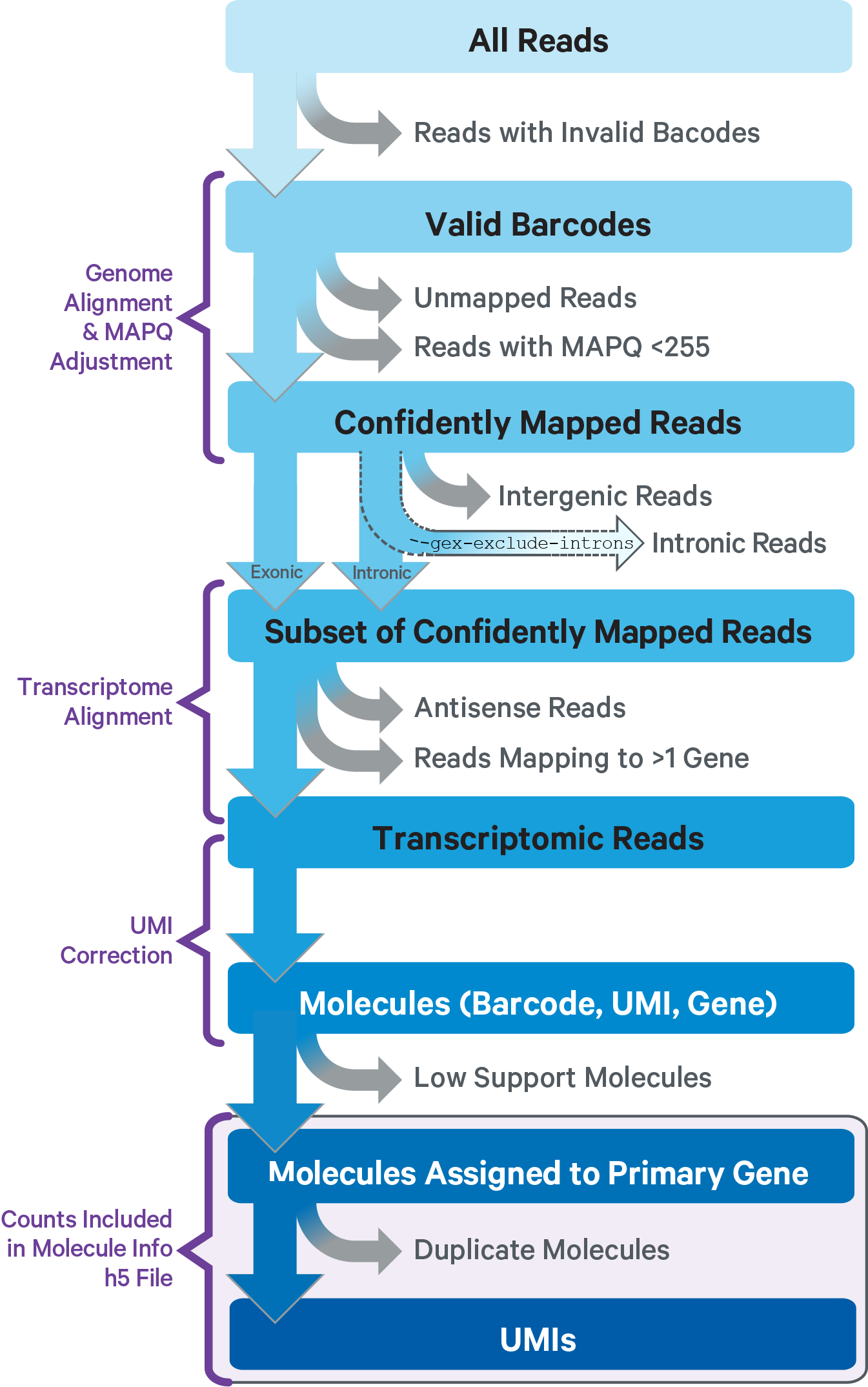
This sub-pipeline performs data analysis to construct the raw GEX count matrix from the input GEX FASTQs and reference. The key read processing steps are outlined in this figure and described in the text below:
A full length cDNA construct is flanked by the 30 bp template switch oligo (TSO) sequence, "AAGCAGTGGTATCAACGCAGAGTACATGGG" on the 5' end and poly-A on the 3' end. Some fraction of sequencing reads are expected to contain either or both these sequences, depending on the library fragment size distribution. Reads derived from short RNA molecules are more likely to contain either or both TSO and poly-A sequence compared to reads derived from longer RNA molecules.
Since the presence of non-template sequence in the form of either template switch oligo (TSO) or poly-A, low-complexity ends confound read mapping, TSO sequence is trimmed from the 5' end of read 2 and poly-A is trimmed from the 3' end prior to alignment. Trimming improves the sensitivity of the assay as well as the computational efficiency of the software pipeline.
Tags ts:i and pa:i in the output BAM files indicate the number of TSO
nucleotides trimmed from the 5' end of read 2 and the number of poly-A
nucleotides trimmed from the 3' end. The trimmed bases are present in the
sequence of the BAM record and are soft clipped in the CIGAR string.
For GEX reads, Cell Ranger ARC uses an aligner called STAR, which peforms splicing-aware alignment of reads to the genome. Cell Ranger ARC then uses the transcript annotation GTF to bucket the reads into exonic, intronic, and intergenic, and by whether the reads align (confidently) to the genome. A read is exonic if at least 50% of it intersects an exon, intronic if it is non-exonic and intersects an intron, and intergenic otherwise.
GEX MAPQ Adjustment: For reads that align to a single exonic locus but also align to 1 or more non-exonic loci, the exonic locus is prioritized and the read is considered to be confidently mapped to the exonic locus with MAPQ 255.
Cell Ranger ARC further aligns exonic and intronic confidently mapped reads to annotated transcripts by examining their compatibility with the transcriptome. As shown below, reads are classified based on whether they are sense or antisense and based on whether they are exonic, intronic or whether their splicing pattern is compatible with transcript annotations associated with that gene.
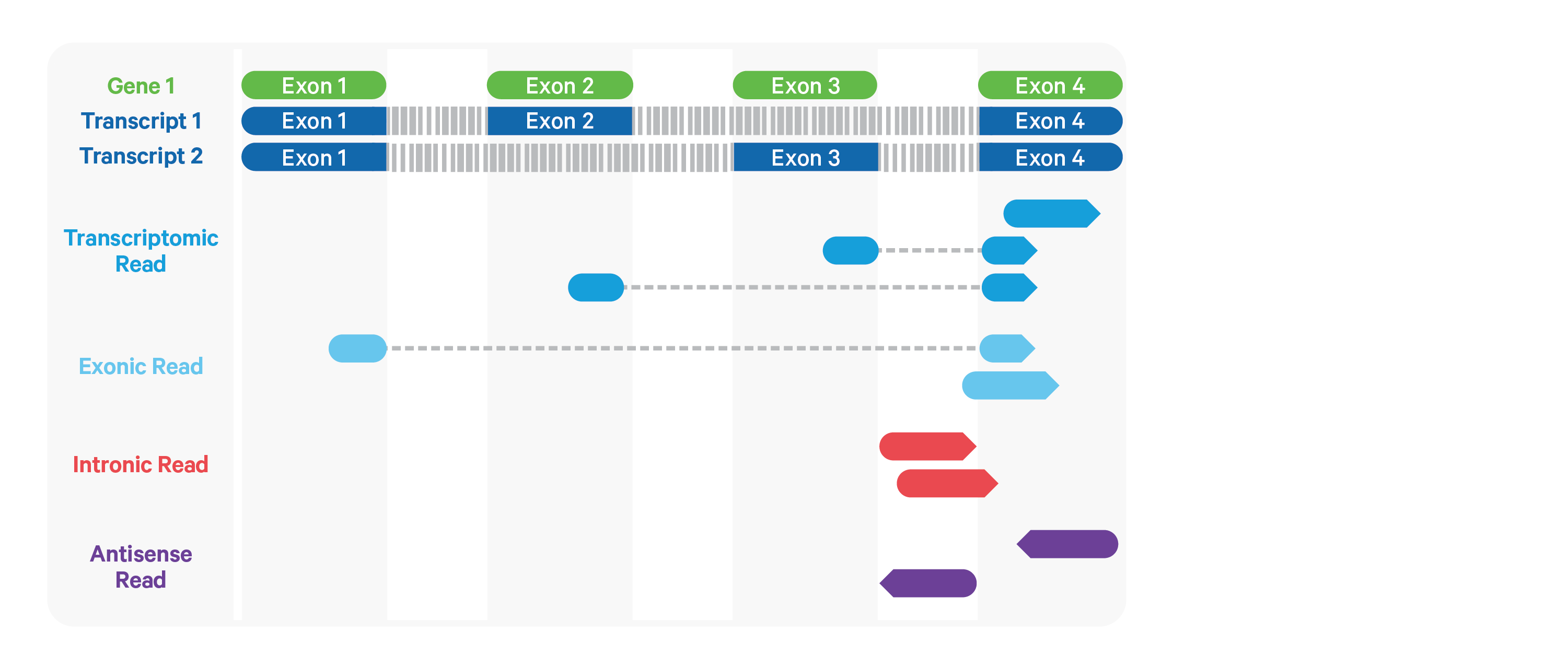
By default, reads that map in the sense orientation to a single gene - which include the reads labeled transcriptomic (blue), exonic (light blue), and intronic (red) in the diagram above - are carried forward to UMI counting. This is because the input to the assay consists of nuclei that are known to be enriched for unspliced transcripts. Users who wish to only count transcriptomic reads (shown in a darker shade of blue above) can do use by running the cellranger-arc count pipeline with the flag --gex-exclude-introns.
|
By default, Cell Ranger ARC includes both exonic and intronic reads that map in the sense orientation to a gene towards UMI counting. This eliminates the need for a special 'nuclei reference' that defines the entire gene body to be an exon. Users can turn off this behavior by using the --gex-exclude-introns flag, in which case only exonic reads that are compatible with annotated splice-junctions are included while counting UMIs. |
Furthermore, a read is considered uniquely mapping if it is compatible with only a single gene. Only uniquely mapping reads are carried forward to UMI counting.
|
Note, in the Web Summary HTML, the set of reads carried forward to UMI counting is referred to as "Reads mapped confidently to transcriptome" |
Before counting UMIs, Cell Ranger ARC attempts to correct for sequencing errors in the UMI sequences. Reads that were confidently mapped to the transcriptome are placed into groups that share the same barcode, UMI, and gene annotation. If two groups of reads have the same barcode and gene, but their UMIs differ by a single base (i.e., are Hamming distance 1 apart), then one of the UMIs was likely introduced by a substitution error in sequencing. In this case, the UMI of the less-supported read group is corrected to the UMI with higher support.
Cell Ranger ARC again groups the reads by barcode, UMI (possibly corrected), and gene annotation. If two or more groups of reads have the same barcode and UMI, but different gene annotations, the gene annotation with the most supporting reads is kept for UMI counting, and the other read groups are discarded. In case of a tie for maximal read support, all read groups are discarded, as the gene cannot be confidently assigned.
After these two filtering steps, each observed barcode, UMI, gene combination is recorded as a UMI count in the raw feature-barcode matrix. The number of reads supporting each counted UMI is also recorded in the molecule info file.
Because of the distinct signal and noise profiles of gene expression and ATAC data, Cell Ranger ARC performs the primary dimensionality reduction on GEX gene-barcode matrix and ATAC peak-barcode matrix using different methods.
In order to reduce the GEX gene-barcode matrix to its most important features, Cell Ranger ARC uses Principal Components Analysis (PCA) to change the dimensionality of the dataset from (cells x genes) to (cells x M) where M is a user-selectable number of principal components (default = 10). The pipeline uses a python implementation of IRLBA algorithm, (Baglama & Reichel, 2005), which we modified to reduce memory consumption.
For ATAC peak-barcode matrix, we normalize the data via the inverse-document frequency (idf) transform, inspired by the large body of work in the field of information retrieval. Each peak count is scaled by the log of the ratio of the number of barcodes in the matrix and the number of barcodes where the peak has a non-zero count. This provides greater weight to counts in peaks that occur in fewer barcodes. Singular value decomposition (SVD) is performed on this normalized matrix using IRLBA without scaling or centering, to produce the transformed matrix in lower dimensional space (default = 15), as well as the components and the singular values signifying the importance of each component. Prior to clustering, we perform normalization to depth by scaling each barcode data point to unit L2-norm in the lower dimensional space. We found that the combination of these normalization techniques obviates the need to remove the first component. Specific to LSA, we provide spherical k-means clustering that produces 2 to 5 clusters for downstream analysis. Spherical k-means was found to perform better than plain k-means, by identifying clusters via k-means on L2-normalized data that lives on the spherical manifold. Similar to PCA, we also provide a graph-based clustering and visualization via t-SNE and UMAP. However, similar to spherical k-means clustering, we normalize the data to unit norm before performing graph-based clustering, t-SNE and UMAP projection.
For visualizing data in 2-d space, Cell Ranger ARC passes the dimensionality reduced data into t-SNE (t-Stochastic Neighbor Embedding), a nonlinear dimensionality reduction method (Van der Maaten, 2014). The C++ reference implementation by Van der Maaten was modified to take a PRNG seed for determinis. We also decreased its runtime by fixing the number of output dimensions at compile time to 2 or 3.
Cell Ranger ARC also supports visualization with UMAP (Uniform Manifold Approximation and Projection), which estimates a topology of the high dimensional data and uses this information to estimate a low dimensional embedding that preserves relationships present in the data. (Leland McInnes et al, 2018) The pipeline uses the python implementation of this algorithm by Leland McInnes.
Cell Ranger ARC uses two different methods for clustering cells by expression similarity, both of which operate in the dimensionality reduced space.
The graph-based clustering algorithm consists of building a sparse nearest-neighbor graph (where cells are linked if they among the k nearest Euclidean neighbors of one another), followed by Louvain Modularity Optimization (LMO; Blondel, Guillaume, Lambiotte, & Lefebvre, 2008), an algorithm which seeks to find highly-connected "modules" in the graph. The value of k, the number of nearest neighbors, is set to scale logarithmically with the number of cells. An additional cluster-merging step is done: Perform hierarchical clustering on the cluster-medoids in the reduced dimension and merge pairs of sibling clusters if there are no genes differentially expressed between them (with B-H adjusted p-value below 0.05). The hierarchical clustering and merging is repeated until there are no more cluster-pairs to merge.
The use of LMO to cluster cells was inspired by a similar method in the R package Seurat
Cell Ranger ARC also performs traditional K-means clustering for K=2,3,4,5, where K is the preset number of clusters.
As peaks are regions enriched for open chromatin, and thus have potential for regulatory function, observing the location of peaks with respect to genes can be insightful. We use bedtools closest -D=b to associate each peak with genes based on closest transcription start sites (packaged within the reference) such that the peak is within 1000 bases upstream or 100 bases downstream of the TSS. Additionally, we also associate genes to putative distal peaks that are much further from the TSS, and are less than 100 kb upstream or downstream from the ends of the transcript (see here for detailed procedures). This association is adopted by our companion visualization software (Loupe Browser) and is used to construct and visualize derived features such as promoter-sums that pool together counts from peaks associated with a gene.
Peaks are enriched for transcription factor (TF) binding motifs and the presence of certain motifs can be indicative of transcription factor activity. To identify these motifs, we first calculate the GC% distribution of peaks and then bin the peaks into equal quantile ranges in the GC content distribution. We use the MOODS Python library packaged inside Cell Ranger ARC to scan each peak for matches to motif position-weight-matrices (PWMs) for transcription factors from the JASPAR database built directly into the reference package. We set a p-value threshold of 1E-7 and background nucleotide frequencies to be the observed nucleotide frequencies within the peak regions in each GC bucket. The list of motif-peak matches is unified across these buckets, thus avoiding GC bias in scanning.
As transcription factors (TF) tend to bind at sites containing their cognate motifs, grouping of accessibility measurements at peaks with common motifs produces a useful enrichment analysis of TFs across single cells. We construct an integer count for each TF for each cell barcode in the following manner: we consider all peaks matched to a given TF, as discovered in the TF motif detection. Then for every barcode, we pool together the cut-site counts across these matched peaks in the filtered peak-barcode matrix. This calculates the total cut-sites in a cell barcode for peaks that share the TF motif. We calculate the proportion of cut-sites for a TF within a barcode out of the total cut-sites for that barcode, which normalizes it to depth. We detect enrichment for a TF by z-scoring the distribution over barcodes of these proportion values for given TF. To make it robust to outliers, we use the modified z-score calculated using the median and the scaled median absolute deviation from the median (MAD), instead of the mean and standard deviation. These z-scored values are visible when you load a dataset in Loupe and accompanies the differential analysis built into Loupe Browser.
| We do not produce the tf-barcode matrix if the motifs.pfm file is missing from the reference package (for example in custom references). We cannot perform TF motif enrichment analysis in these cases. |
In order to identify genes whose expression is specific to each cluster, Cell Ranger ARC tests whether the in-cluster mean differs from the out-of-cluster mean for each gene and each cluster.
In order to find differentially expressed genes between groups of cells, Cell Ranger ARC uses the quick and simple method sSeq (Yu, Huber, & Vitek, 2013), which employs a negative binomial exact test. When the counts become large, Cell Ranger ARC switches to the fast asymptotic beta test used in edgeR (Robinson & Smyth, 2007). For each cluster, the algorithm is run on that cluster versus all other cells, yielding a list of genes that are differentially expressed in that cluster relative to the rest of the sample.
Cell Ranger ARC's implementation differs slightly from that in the paper: in the sSeq paper, the authors recommend using DESeq's geometric mean-based definition of library size. Cell Ranger ARC instead computes relative library size as the total UMI counts for each cell divided by the median UMI counts per cell. As with sSeq, normalization is implicit in that the per-cell library-size parameter is incorporated as a factor in the exact-test probability calculations.
In order to identify transcription factor motifs whose accessibility is specific to each cluster, Cell Ranger ARC tests, for each motif and each cluster, whether the in-cluster mean differs from the out-of-cluster mean. Users familiar with our Chromium Single Cell Gene Expression Solution may recognize that this is identical to what Cell Ranger ARC does for identifying differential gene expression. In order to find differentially accessible motifs between groups of cells, Cell Ranger ARC uses a Negative Binomial (NB2) generalized linear model to discover the cluster specific means and their standard deviations, and then employs a Wald test for inference. For each cluster, the algorithm is run on that cluster versus all other cells, yielding a list of TF motifs that are differentially expressed in that cluster relative to the rest of the sample. Using a GLM framework allows us to model the sequencing depth per cell and GC content in peaks per cell directly as covariates. This allows us to normalize to them naturally as part of model estimation and inference procedure. We also perform differential enrichment analysis for accessibility in peaks using a Poisson generalized linear model, much the same way as we do for TF motifs. In this case, we model only the per cell depth as a covariate.
| We cannot perform differential analysis for transcription factor motifs in cases where the motifs.pfm file is missing from the reference package, such as in custom references built without the motif file. |
Baglama, J. & Reichel, L. Augmented Implicitly Restarted Lanczos Bidiagonalization Methods. SIAM Journal on Scientific Computing 27, 19–42 (2005).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment 2008, (2008).
McInnes, L, Healy, J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018
Robinson, M. D. & Smyth, G. K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332 (2007). Link to edgeR source.
Van der Maaten, L.J.P. Accelerating t-SNE using Tree-Based Algorithms. Journal of Machine Learning Research 15, 3221-3245 (2014).
Yu, D., Huber, W. & Vitek, O. Shrinkage estimation of dispersion in Negative Binomial models for RNA-seq experiments with small sample size. Bioinformatics 29, 1275–1282 (2013).