Cell Ranger ARC1.0, printed on 04/06/2025
Cluster mode is one of three primary ways of running Cell Ranger ARC. To learn about the other approaches, see the computing options page.
Cell Ranger ARC can be run in cluster mode, using LSF, SGE, or Slurm to run the stages on multiple nodes via batch scheduling. This allows highly parallel stages to utilize hundreds or thousands of cores concurrently, thereby significantly reducing time to solution.
| 10x does not officially support Torque/PBS. Some customers have successfully used Cell Ranger ARC with those job schedulers in cluster mode. However, it is unsupported and may require trial and error attempts. |
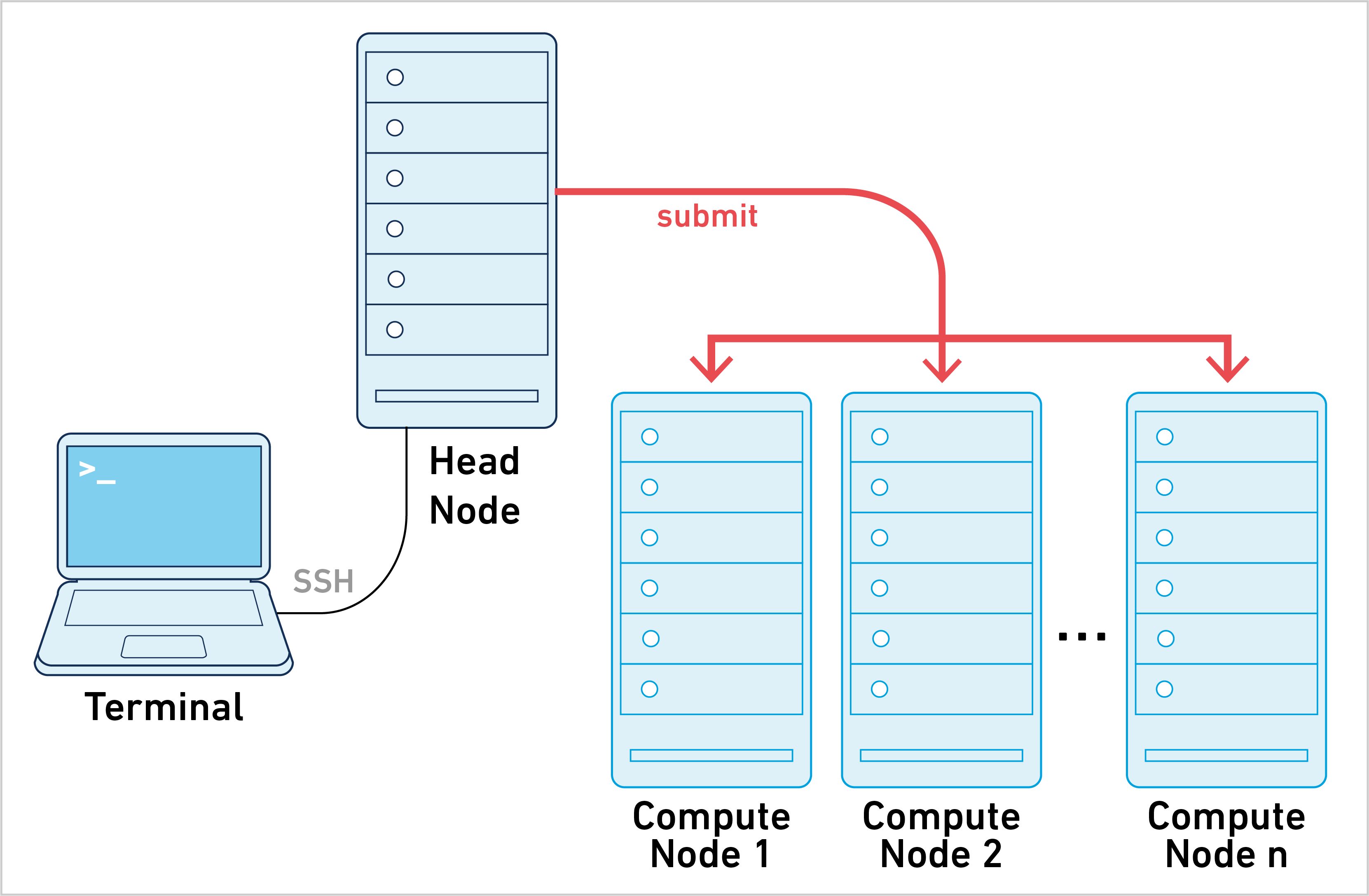
Running pipelines in cluster mode requires the following:
Installing the Cell Ranger ARC software on a cluster is identical to installation on a local server. After confirming that the cellranger-arc commands can run in single server mode, configure the job submission template that Cell Ranger ARC uses to submit jobs to the cluster. Assuming Cell Ranger ARC is installed to /opt/cellranger-arc-1.0.1, the process is as follows:
Step 1. Navigate to the Martian runtime's jobmanagers/ directory which contains example jobmanager templates.
$ cd /opt/cellranger-arc-1.0.1/external/martian/jobmanagers $ ls config.json lsf.template.example sge.template.example slurm.template.example
Step 2. Make a copy of the cluster's example template (LSF, SGE, or Slurm) to either lsf.template, sge.template or slurm.template in the jobmanagers/ directory.
$ cp -v sge.template.example sge.template `sge.template.example' -> `sge.template' $ ls config.json lsf.template.example sge.template sge.template.example slurm.template.example
The job submission templates contain a number of special variables that are substituted by the Martian runtime when each stage is submitted. Specifically, the following variables are expanded when a pipeline is submitting jobs to the cluster:
| Variable | Must be present? | Description |
|---|---|---|
| __MRO_JOB_NAME__ | Yes | Job name composed of the sample ID and stage being executed |
| __MRO_THREADS__ | No | Number of threads required by the stage |
| __MRO_MEM_GB__ __MRO_MEM_MB__ |
No | Amount of memory (in GB or MB) required by the stage |
| __MRO_MEM_GB_PER_THREAD__ __MRO_MEM_MB_PER_THREAD__ |
No | Amount of memory (in GB or MB) required per thread in multi-threaded stages. |
| __MRO_STDOUT__ __MRO_STDERR__ |
Yes | Paths to the _stdout and _stderr metadata files for the stage |
| __MRO_CMD__ | Yes | Bourne shell command to run the stage code |
It is critical that the special variables listed as required are present in the final template you create. For more information on how the template should appear for a cluster, consult your cluster administrator or help desk.
Depending on which job scheduler you have, please select a tab below.
The most common modifications to the job submission template include adding additional lines to specify:
To run a Cell Ranger ARC pipeline in cluster mode, add one of --jobmode=lsf, --jobmode=sge or --jobmode=slurm command-line option when using the cellranger-arc commands. It is also possible to use --jobmode=<PATH>, where <PATH> is the full path to the cluster template file.
To validate that cluster mode is properly configured, follow the same validation instructions given for cellranger-arc in the Installation page and specify the appropriate cluster via --jobmode.
$ cellranger-arc mkfastq --run=./tiny-bcl --samplesheet=./tiny-sheet.csv --jobmode=sge Martian Runtime - 1.0.1 Running preflight checks (please wait)... 2016-09-13 12:00:00 [runtime] (ready) ID.HAWT7ADXX.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET 2016-09-13 12:00:00 [runtime] (split_complete) ID.HAWT7ADXX.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET ...
Once the preflight checks are finished, check the job queue to see stages queuing up:
$ qstat job-ID prior name user state submit/start at queue slots ja-task-ID ----------------------------------------------------------------------------------------------------------------- 8675309 0.56000 ID.HAWT7AD jdoe qw 09/13/2016 12:00:00 all.q@cluster.university.edu 1 8675310 0.55500 ID.HAWT7AD jdoe qw 09/13/2016 12:00:00 all.q@cluster.university.edu 1
In the event of a pipeline failure, an error message is displayed:
[error] Pipestance failed. Please see log at: HAWT7ADXX/MAKE_FASTQS_CS/MAKE_FASTQS/MAKE_FASTQS_PREFLIGHT/fork0/chnk0/_errors Saving diagnostics to HAWT7ADXX/HAWT7ADXX.debug.tgz For assistance, upload this file to 10x by running: uploadto10x <your_email> HAWT7ADXX/HAWT7ADXX.debug.tgz
The _errors file contains a jobcmd error:
$ cat HAWT7ADXX/MAKE_FASTQS_CS/MAKE_FASTQS/MAKE_FASTQS_PREFLIGHT/fork0/chnk0/_errors jobcmd error: exit status 1
The most likely reason for this failure is an invalid job submission template. This occurs when the job submission via bsub, qsub, or sbatch commands failed.
The "tiny" dataset does not stress the cluster, so it is worthwhile to follow up with a more realistic test using one of our sample datasets
There are two subtle variants of running cellranger-arc pipelines in cluster mode, each with their own pitfalls. Check with your cluster administrator to see which approach is compatible with your institution's setup.
When cellranger-arc is run in cluster mode, a single library analysis is partitioned into hundreds and potentially thousands of smaller jobs. The underlying Martian pipeline framework launches each stage job using the bsub, qsub or sbatch commands when running on LSF, SGE, or Slurm cluster, respectively. As stage jobs are queued, launched, and completed, the pipeline framework tracks their status using the metadata files that each stage maintains in the pipeline output directory.
Like single server pipelines, cluster-mode pipelines can be restarted after a failure. They maintain the same order of execution for the dependent stages of the pipeline. All executed stage code is identical to single server mode and the quantitative results are identical to the limit of each stage's reproducibility.
| Cluster-mode pipelines that are stopped (either by you or due to a stage failure) do not delete pending stages that have already been submitted to the cluster queue. As a result, some pipeline stages may continue to execute after the cellranger-arc commands have exited. |
In addition, the Cell Ranger ARC UI can still be used with cluster mode. Because the Martian pipeline framework runs on the node from which the command was issued, the UI will also run from that node.
Stages in the Cell Ranger ARC pipelines each request a specific number of cores and memory to aid with resource management. These values are used to prevent oversubscription of the computing system when running pipelines in single server mode. The way CPU and memory requests are handled in cluster mode is defined by the following:
Depending on which job scheduler you have, select a tab below:
$ cat bsub.template #BSUB -J __MRO_JOB_NAME__ #BSUB -n __MRO_THREADS__ #BSUB -o __MRO_STDOUT__ #BSUB -e __MRO_STDERR__ #BSUB -R "rusage[mem=__MRO_MEM_MB__]" #BSUB -R span[hosts=1] __MRO_CMD__
$ cat sge.template #$ -N __MRO_JOB_NAME__ #$ -V #$ -pe threads __MRO_THREADS__ #$ -l mem_free=__MRO_MEM_GB__G #$ -cwd #$ -o __MRO_STDOUT__ #$ -e __MRO_STDERR__ __MRO_CMD__
| In the above example, the trailing G in the highlighted __MRO_MEM_GB__G is required by SGE to denote that mem_free is being expressed in GB units. |
Note that the h_vmem (virtual memory) and mem_free/h_rss (physical memory) represent two different quantities and that Cell Ranger ARC stages' __MRO_MEM_GB__ requests are expressed as physical memory. Using h_vmem in your job template may cause certain stages to be unduly killed if their virtual memory consumption is substantially larger than their physical memory consumption. It follows that we do not recommend using h_vmem.
| If you do use h_vmem in a template, it is recommended that you use the __MRO_MEM_GB_PER_THREAD__ or __MRO_MEM_MB_PER_THREAD__ variables instead of __MRO_MEM_GB__ and __MRO_MEM_MB__. To determine memory limits for a multicore job, SGE will multiply the number of threads by the value in h_vmem. The __MRO_MEM_GB__ and __MRO_MEM_MB__ variables already reflect the sum amount of memory across all threads needed to run the job. Using those variables as h_vmem will inflate the memory required for multi-threaded jobs. |
$ cat slurm.template #SBATCH -J __MRO_JOB_NAME__ #SBATCH --export=ALL #SBATCH --nodes=1 --ntasks-per-node=__MRO_THREADS__ #SBATCH --signal=2 #SBATCH --no-requeue ### Alternatively: --ntasks=1 --cpus-per-task=__MRO_THREADS__ ### Consult with your cluster administrators to find the combination that ### works best for single-node, multi-threaded applications on your system. #SBATCH --mem=__MRO_MEM_GB__G #SBATCH -o __MRO_STDOUT__ #SBATCH -e __MRO_STDERR__ __MRO_CMD__
For clusters whose job managers do not support memory requests, it is possible to request memory in the form of cores via the --mempercore command-line option. This option scales up the number of threads requested via the __MRO_THREADS__ variable according to how much memory a stage requires.
For example, given a cluster with nodes that have 16 cores and 128 GB of memory (8 GB per core), the following pipeline invocation command
$ cellranger-arc mkfastq --run=./tiny-bcl --samplesheet=./tiny-sheet.csv --jobmode=sge --mempercore=8
will issue the following resource requests:
As the final bullet point illustrates, this mode can result in wasted CPU cycles and is only provided for clusters that cannot allocate memory as an independent resource.
Every cluster configuration is different. If you are unsure of how your cluster resource management is configured, contact your cluster administrator or help desk.
Some Cell Ranger ARC pipeline stages are divided into hundreds of jobs. By default, the rate at which these jobs are submitted to the cluster is throttled to at most 64 at a time and at least 100ms between each submission to avoid running into limits on clusters which impose quotas on the total number of pending jobs a user can submit.
If your cluster does not have these limits or is not shared with other users, you can control how the Martian pipeline runner sends job submissions to the cluster by using the --maxjobs and --jobinterval parameters.
To increase the cap on the number of concurrent jobs to 200, use the --maxjobs parameter:
$ cellranger-arc count --id=sample ... --jobmode=sge --maxjobs=200
You can also change the rate limit on how often the Martian pipeline runner sends submissions to the cluster. To add a five-second pause between job submissions, use the --jobinterval parameter:
$ cellranger-arc count --id=sample ... --jobmode=sge --jobinterval=5000
The job interval parameter is in milliseconds. The minimum allowable value is 1.