Cell Ranger DNA1.1, printed on 03/29/2025
| Analysis software for the 10x Genomics single cell DNA product is no longer supported. Raw data processing pipelines and visualization tools are available for download and can be used for analyzing legacy data from 10x Genomics kits in accordance with our end user licensing agreement without support. |
For an approximately diploid human sample we recommend a sequencing depth of 750,000 read-pairs per cell. At this depth, the metric median effective reads per 1Mbp is between 350-400, and we expect to be able to detect single cell copy number events in the size range 1-2 megabases (and upwards) with high sensitivity and positive predictive value. In groups of 10 or more cells we expect to be able to detect copy number events in the 100-200 kilobase (and upwards) with high sensitivity and positive predictive value.
Sequencing below the recommended depth will result in lower sensitivity to small copy number events. The metric median effective_reads per 1Mbp is a good predictor of CNV calling performance rather than the total number of sequencing reads obtained. Shown below is the variation in sensitivity of event detection as a function of the median effective_reads per 1Mbp for different event sizes calculated by downsampling a dataset of MKN-45 cells.
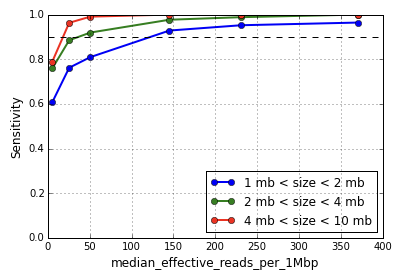
Note that the performance degrades gently with reduced read depth and falls off sharply below median effective reads per 1Mbp of 50 corresponding to an input of approximately 200,000 reads per cell. The algorithms have not been extensively tested at sequencing depths below 200,000 reads per cell and the results are potentially unreliable.
We expect high quality CNV detection when median effective reads per 1Mbp is in the range 350-400, and the results in the graph above will likely translate across organisms. This level of coverage can be achieved by scaling the recommended coverage of 1.5-2.0 million reads per cell by the ratio of the organism genome size to the human genome size.
For samples that contain cells with average ploidy significantly different from two, as is some times the case in cancer genomes, we recommend scaling the input coverage in proportion to the average ploidy / 2. In a tetraploid sample, for example, the extra coverage allows us to distinguish 4 -> 5 copy number changes and other n -> n+1 higher copy number transitions where the relative ploidy difference can be small.
As the sequencing depth per cell is reduced the uncertainty in the start and end of a copy number event increases. This increases the clustering distance between a pairs of cells and the structure of the hierarchical clustering can be affected as a consequence. For example, consider a hypothetical sample with 3 cells, where cells 1 and 2 have identical copy number profiles, and cell 3 has an additional 10 megabase copy number event. If the sequencing depth is drastically reduced, these three cells could appear equidistant from each other because of inaccuracies in precisely localizing the start/end of the event.