Cell Ranger3.0, printed on 04/01/2025
While most of the antibody-related metrics and counts are computed in parallel to their gene expression counterparts, there are some unique aspects of protein libraries that specific analysis steps in Cell Ranger 3.0.
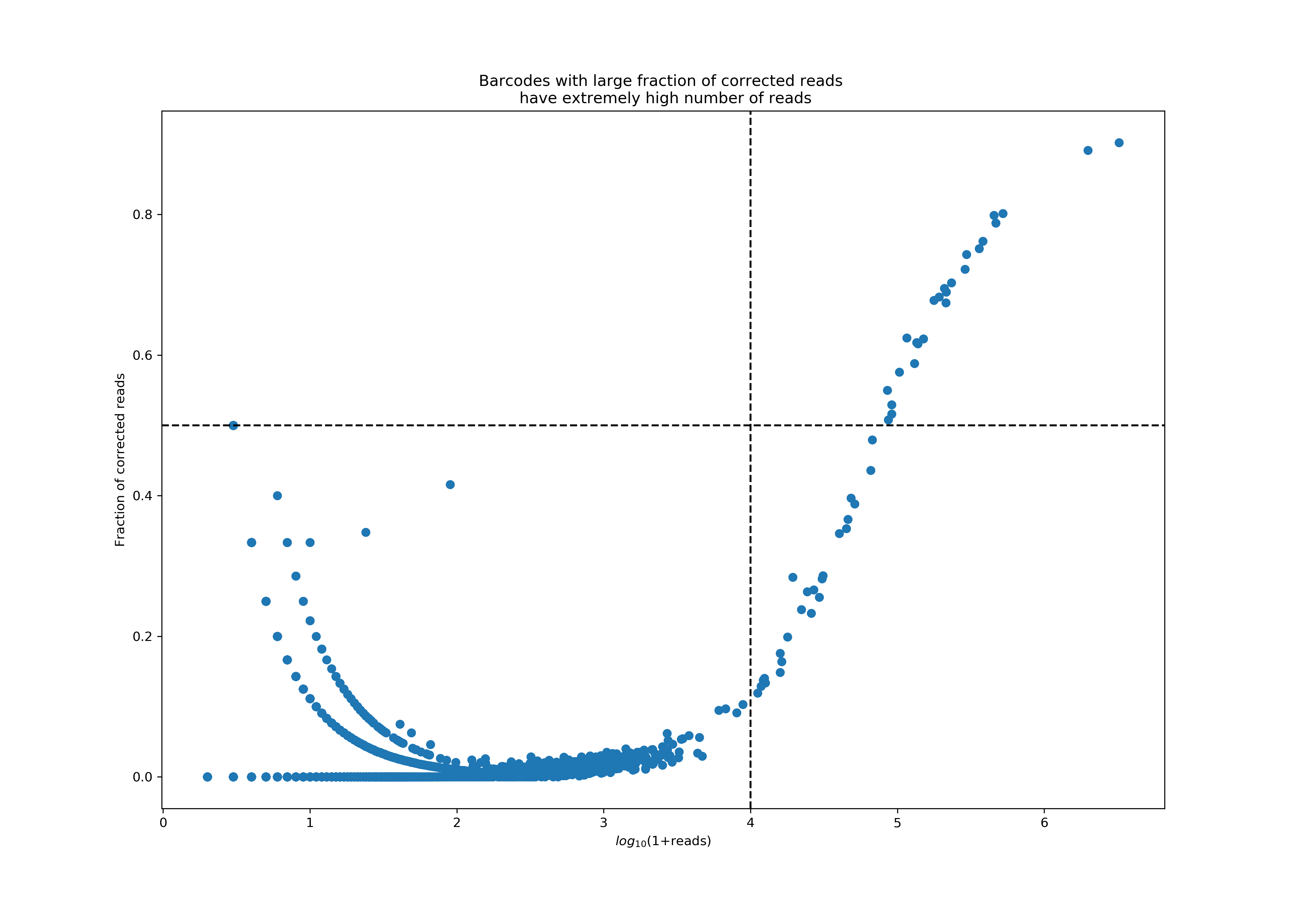
Protein aggregates in antibody staining reagents cause a few GEMs to have extremely high UMI counts. As part of the UMI counting pipeline, Cell Ranger looks for pairs of UMIs that are different only in one base (i.e., are 1-Hamming distance away) and implements a UMI correction step, during which these reads will be combined into a single UMI count by correcting the UMI with fewer reads into the UMI with more reads. While such correction events are typically rare, we observed that in Antibody libraries sometimes the corrections rates are abnormally high, a phenomenon that always correlates with extremely high UMI counts. These high UMI counts will cause saturation of the UMI space, leading to false UMI corrections. Protein aggregates are one leading cause of such high UMI counts and correction rates.
The plot below shows a particularly bad example of protein aggregation, where a handful of barcodes accounted for almost 77% of all reads, with extremely high correction rates in these reads. Currently, we consider a barcode an aggregate if it has more than 10K reads, 50% of which were corrected. Cell Ranger 3.0 will flag such barcodes and remove them from the final feature-barcode matrix, and report the fraction of reads associated with such barcodes in the Antibody: Fraction Reads in Barcodes with High UMI Counts metric on the web summary. In addition, two more related metrics will be available from metrics_summary.json; Antibody: Number of Barcodes with High UMI Correction Rate is the number of detected aggregate barcodes, and Antibody: Fraction Reads that have Corrected UMIs is the fraction of all reads that have a correction event.
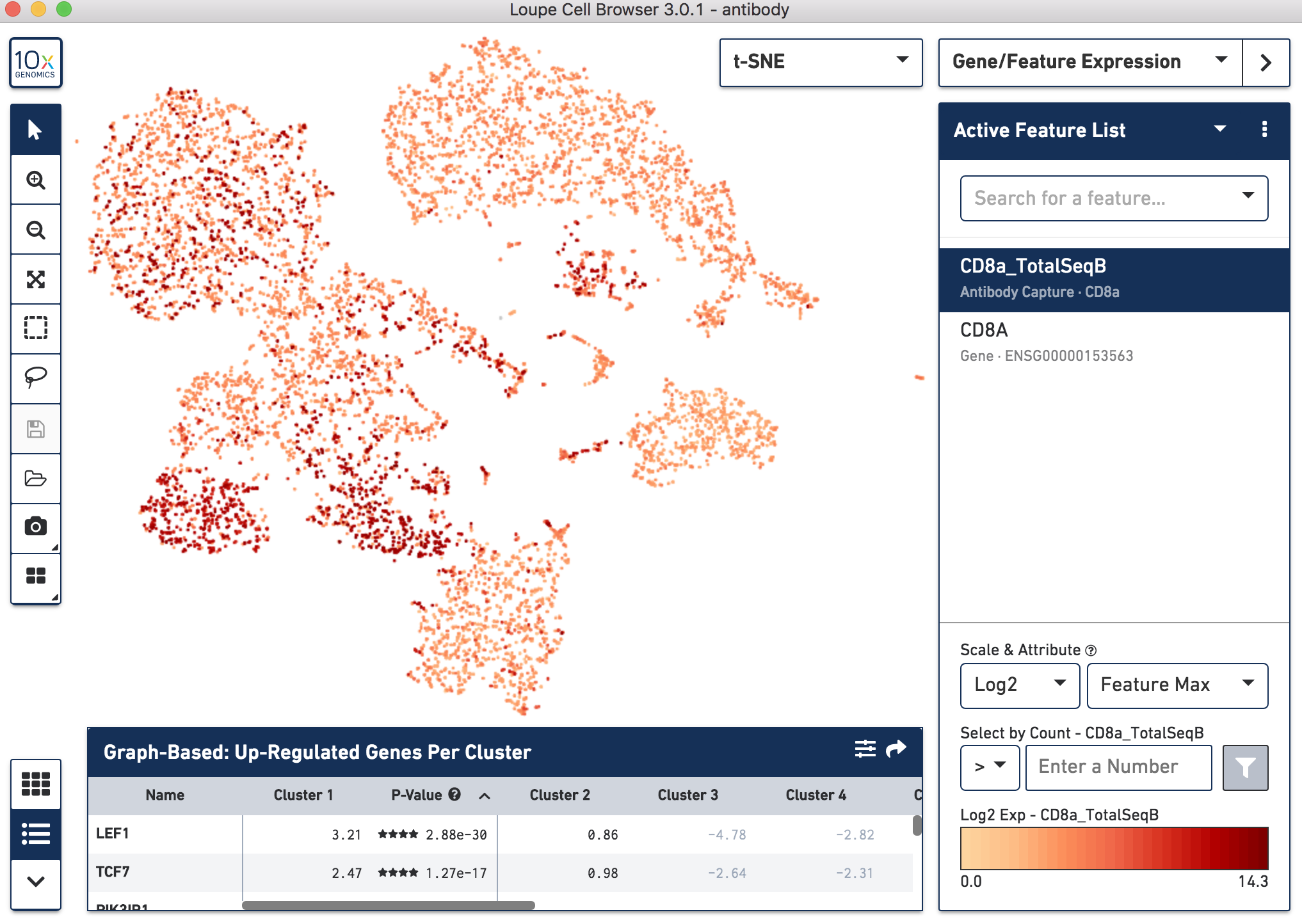
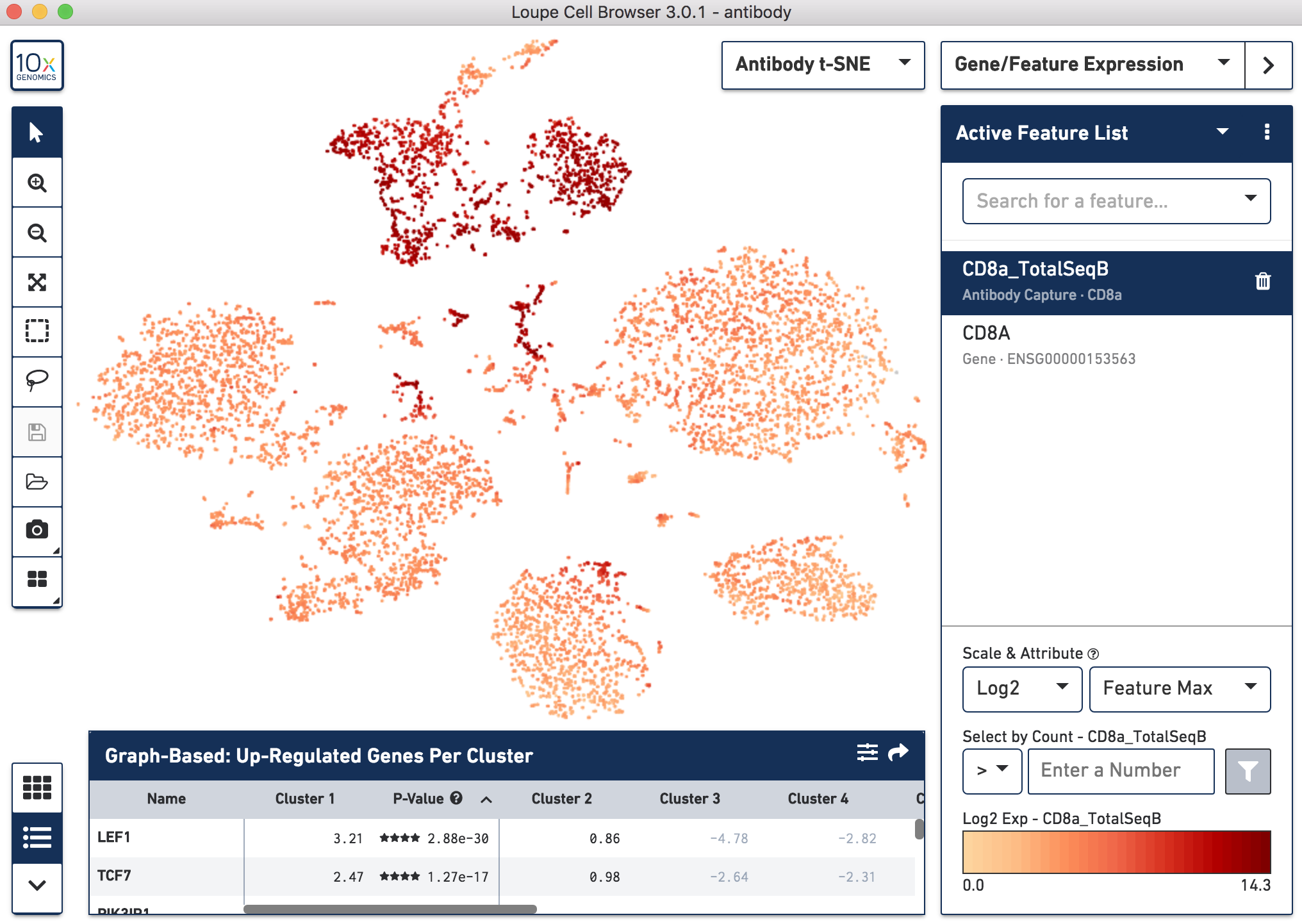
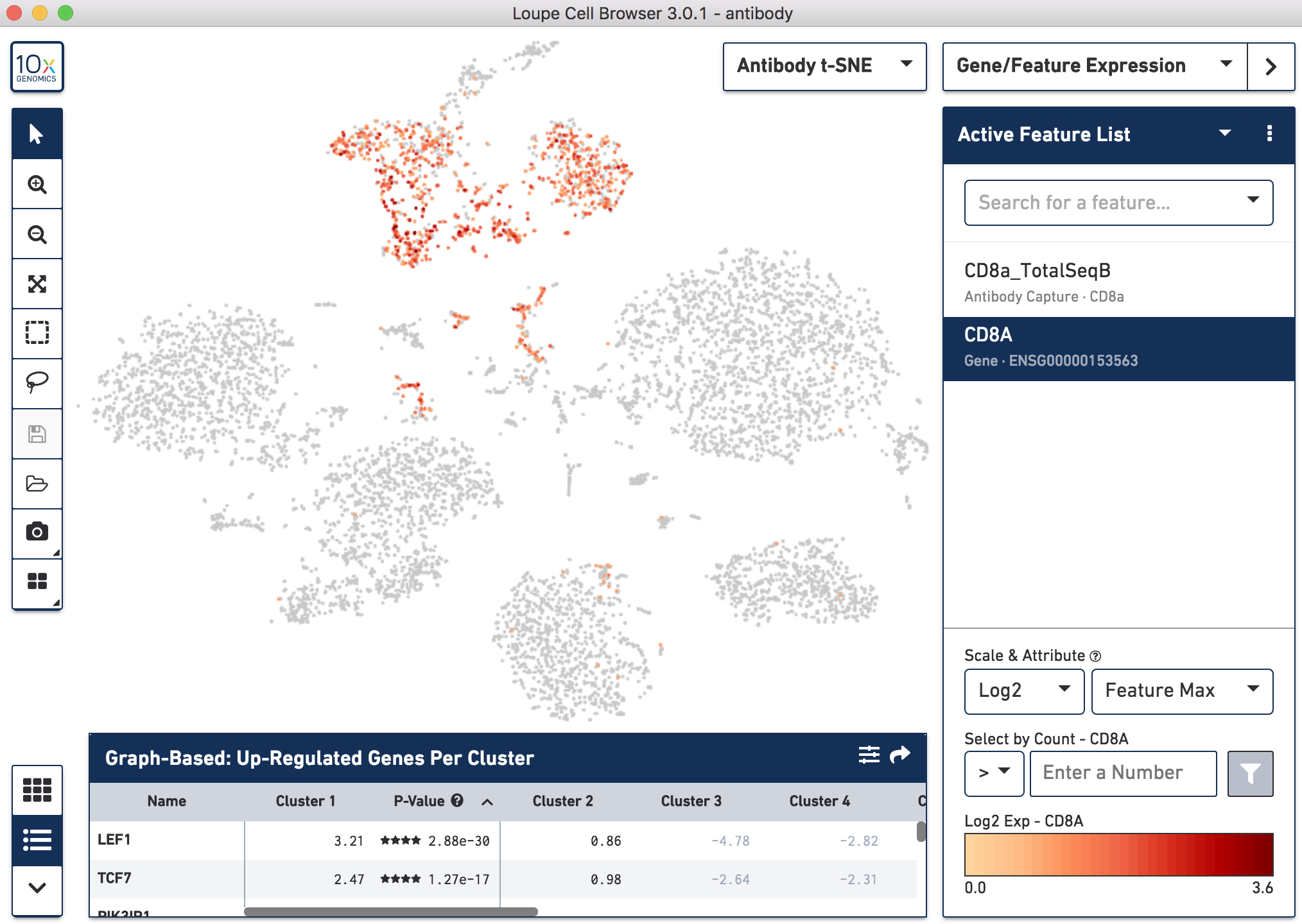
Since cell-surface proteins offer a unique and complementary view of the cell types on top of the genes they express, Cell Ranger 3.0 runs a popular t-distributed Stochastic Neighbor Embedding (t-SNE) algorithm to visualize the protein counts in 2-D space. On samples where Antibody Capture is an input library, the pipeline will slice out the feature counts from the full matrix and perform a t-SNE on these raw counts (unlike the gene expression part of the matrix, where t-SNE is run on the PCA-reduced space). These t-SNE projections then can be visualized with the Loupe Cell Browser versions 3.0 and above.
Below, the left panel shows a traditional t-SNE with gene counts only, overlaying the counts of CD8a antibody. The middle panel shows CD8a protein expression overlayed on t-SNE projections computed on antibody counts only. The right panel shows the expression of CD8A gene on the antibody-derived t-SNE projections for comparison.
 |
 |
 |