After uploading input files into your project in Cloud Analysis, you are ready to analyze your data.
The FASTQ files uploaded into your project have automatically been grouped into sets upon upload. To determine which pipeline is appropriate for your data, you must specify the library or feature type corresponding to the FASTQ sets that you are analyzing. In this example, we analyze a Visium HD mouse kidney dataset within a project titled Visium HD.
You may assign values one at a time using the dropdown selectors contained in each row of the table, highlighted in the screenshot below.
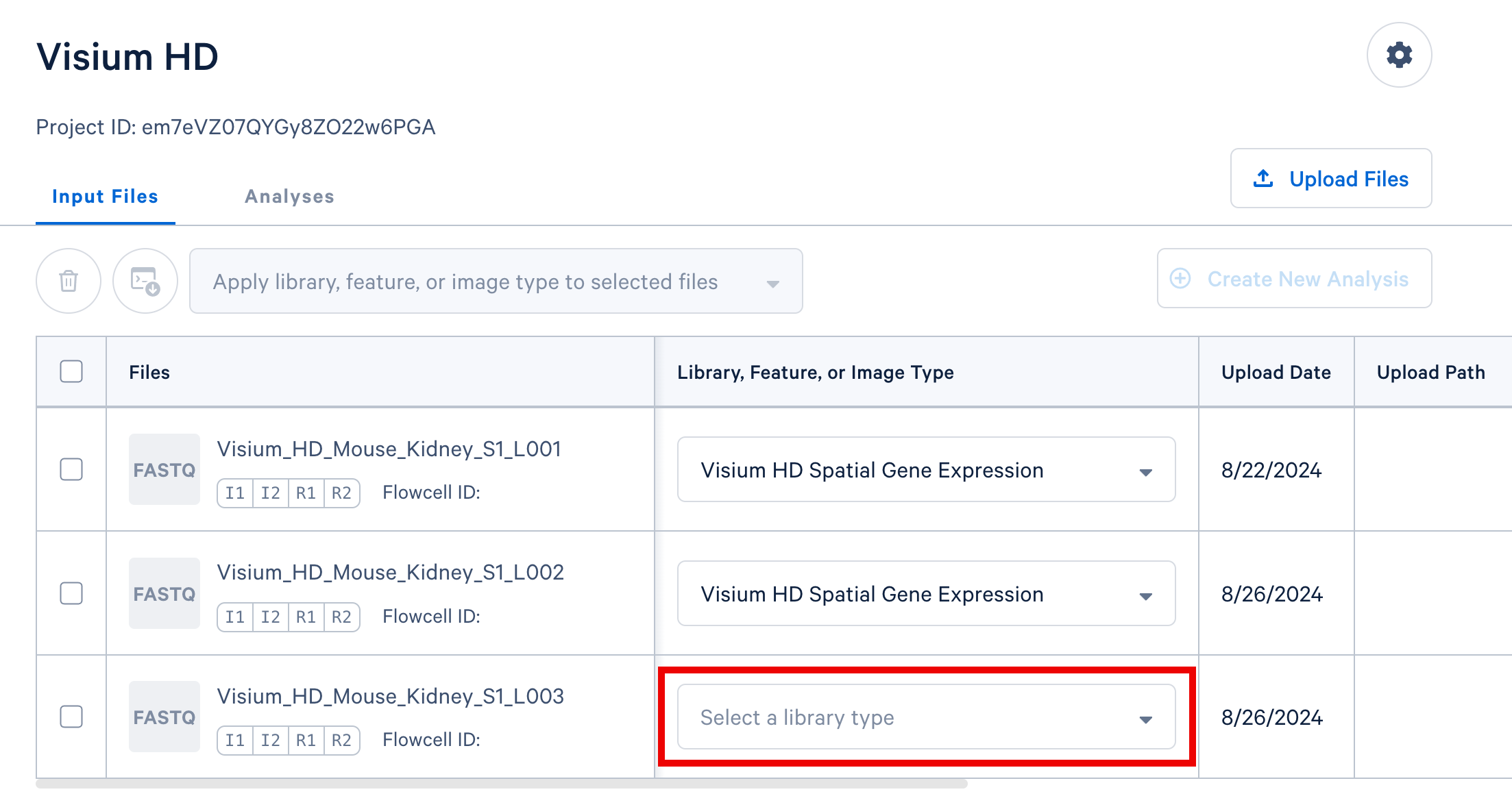
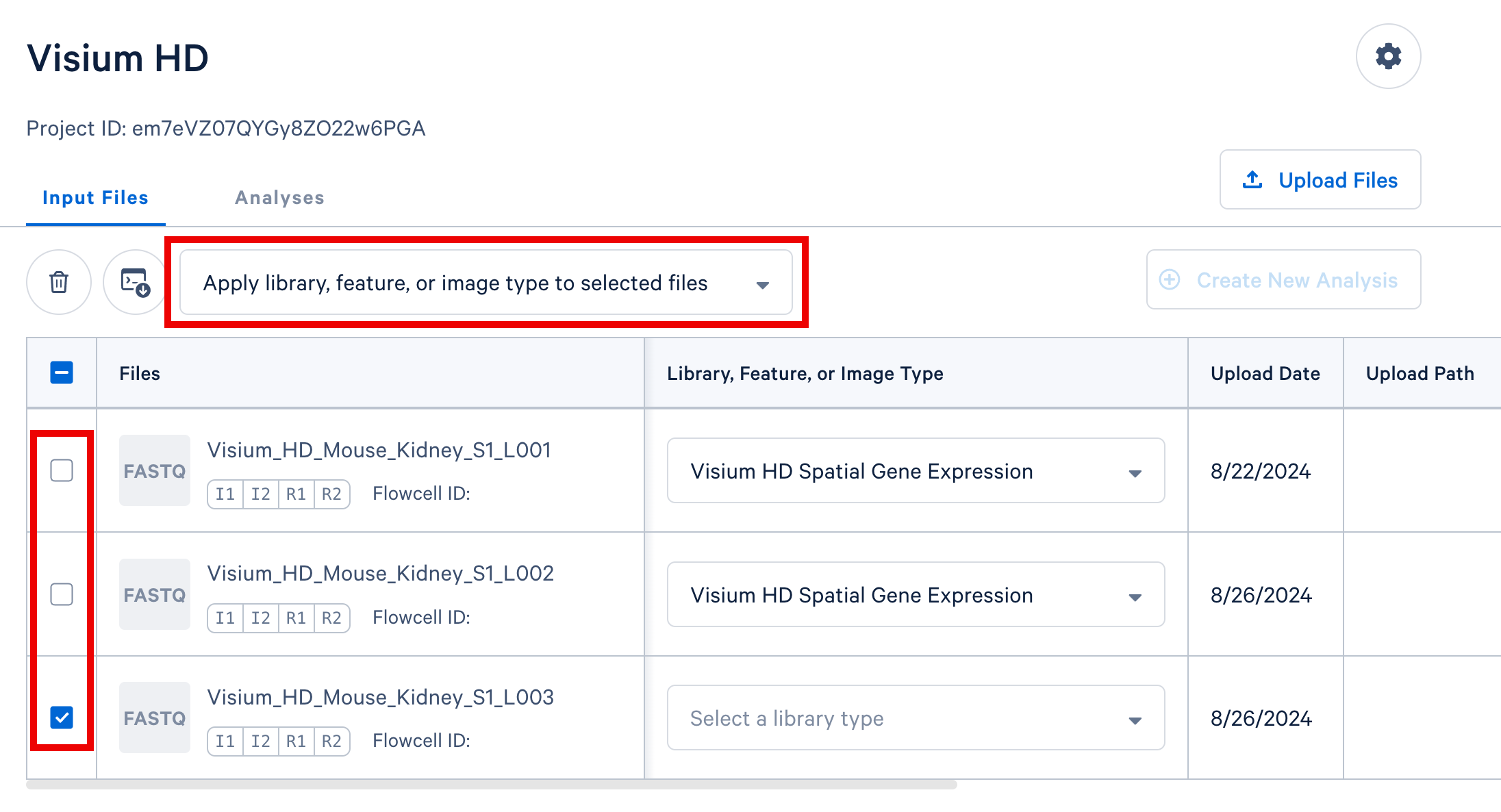
If you have multiple FASTQ sets that share a library or feature type, you may select these rows in the table and apply a single library or feature type value to all selected rows using the selector that appears above the FASTQ file table, highlighted in the screenshot below.
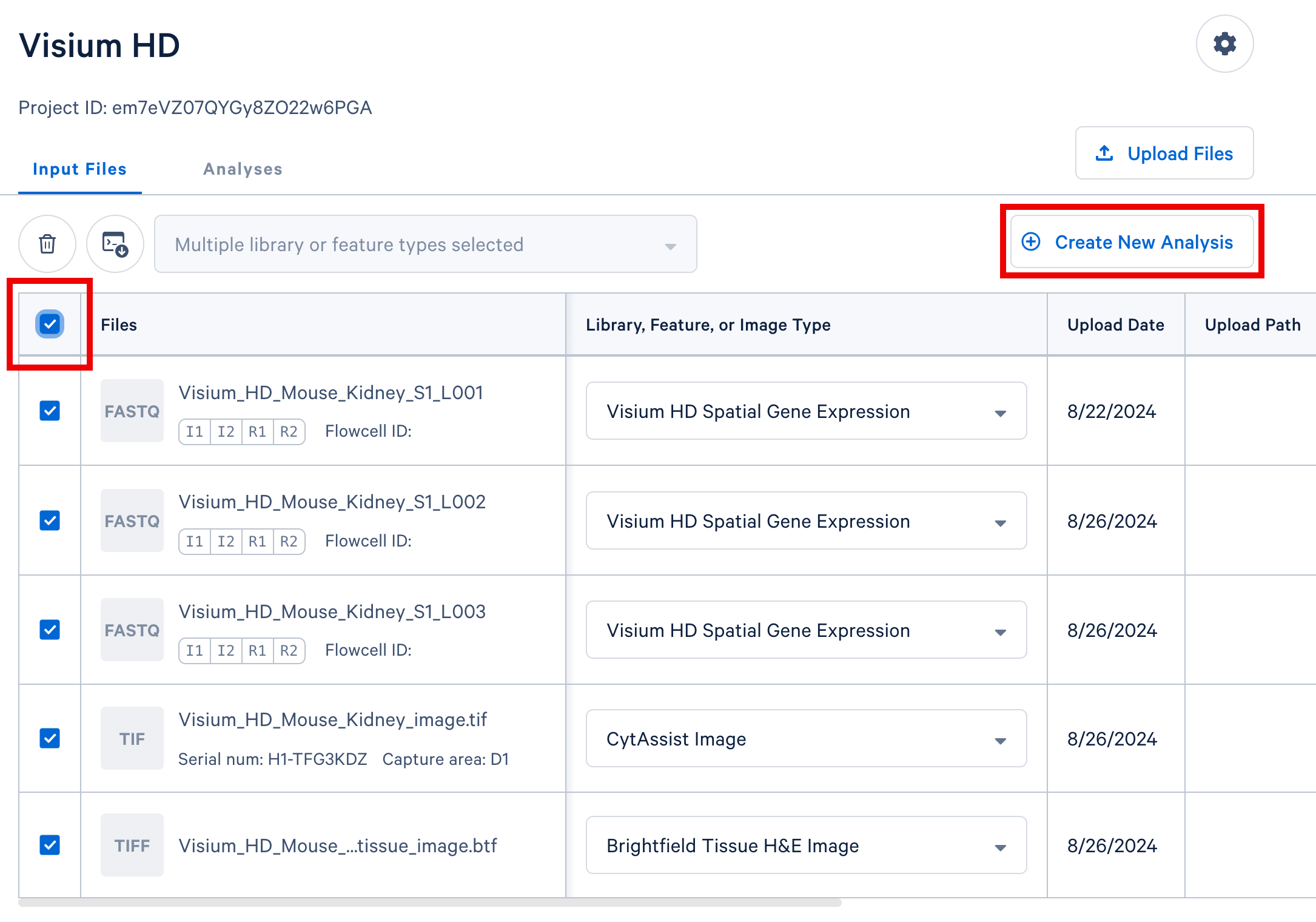
In your project in Cloud Analysis, select the input files from a single experiment (either a single GEM well for single cell, or a single slide for spatial) and click the Create New Analysis button to proceed. In this example, we include three sets of FASTQ files from three lanes on the sequencer, the CytAssist image, and the brightfield H&E tissue image.
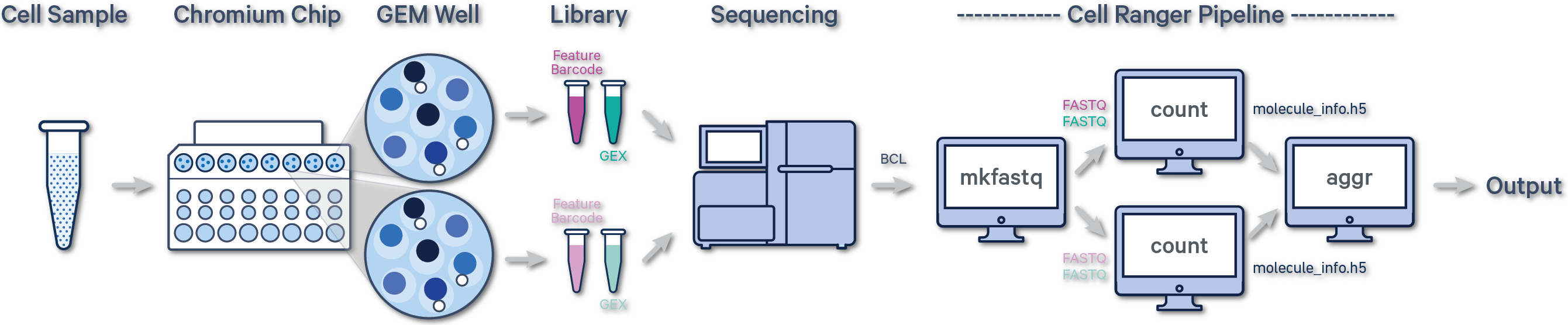
The Cell Ranger and Cell Ranger ARC pipelines, except for the corresponding aggr pipelines, expect FASTQ files from a single GEM well. One or more sequencing libraries can be derived from a GEM Well. Additionally, one or more FASTQ sets can be generated for each sequencing library depending on the sequencing configuration. More details and examples can be found in the Cell Ranger documentation for Single Cell Gene Expression and Immune Profiling data or the Cell Ranger ARC documentation for Multiome data.
Depending on the types of data that are being analyzed, we will automatically open a setup form corresponding to our recommended pipeline for your data. By default, the newest versions are recommended. You may select older versions from the “Pipeline” field in the setup form.
Each field has a short description to explain the appropriate usage. If you need more information about any field, please consult the appropriate documentation:
- Space Ranger count for Visium HD data
- Cell Ranger documentation for Single Cell Gene Expression and Immune Profiling data
- Cell Ranger ARC documentation for Multiome data
After entering values for all required fields and the appropriate optional fields, the “Run Analysis” button will be selectable. Clicking this button will initiate your pipeline run. Compute time varies based on the size and types of data that you are analyzing, but typically runs take a few hours to complete. By default, you will receive an email notification when your analyses complete. If you would like to disable this notification, please update your preferences on your Account Settings.
After your analysis completes, you can download your output files. Learn more about downloading outputs.
For experiments with data from multiple GEM wells, each library must be analyzed with the Cell Ranger count pipeline first and then aggregated with the aggr pipeline.
(A single GEM well can yield multiple physical libraries: one Gene Expression library and one or more Feature Barcode libraries. The example below has one Gene Expression library per GEM well.)
Here, 1k peripheral blood mononuclear cell (PBMC) and 10k PBMC datasets were each analyzed as separate runs of the Cell Ranger count pipeline. Select the FASTQ files for a given library, click Create New Analysis, and follow the setup form steps (e.g., as shown in this video guide).
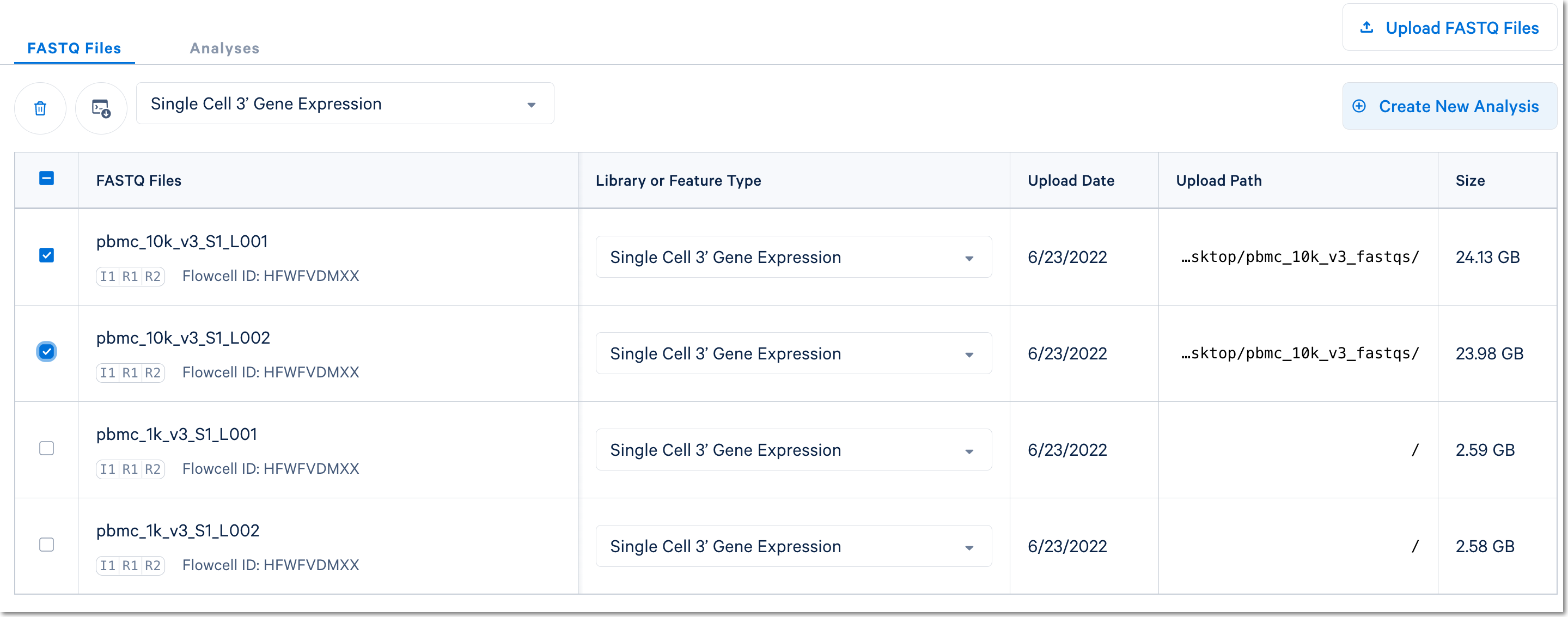
(Note: in the screenshot above, the Upload Path is blank for files uploaded with the Web Browser "Browse Files" option)
Once the count analyses are complete for each library, the aggregation option will be visible:
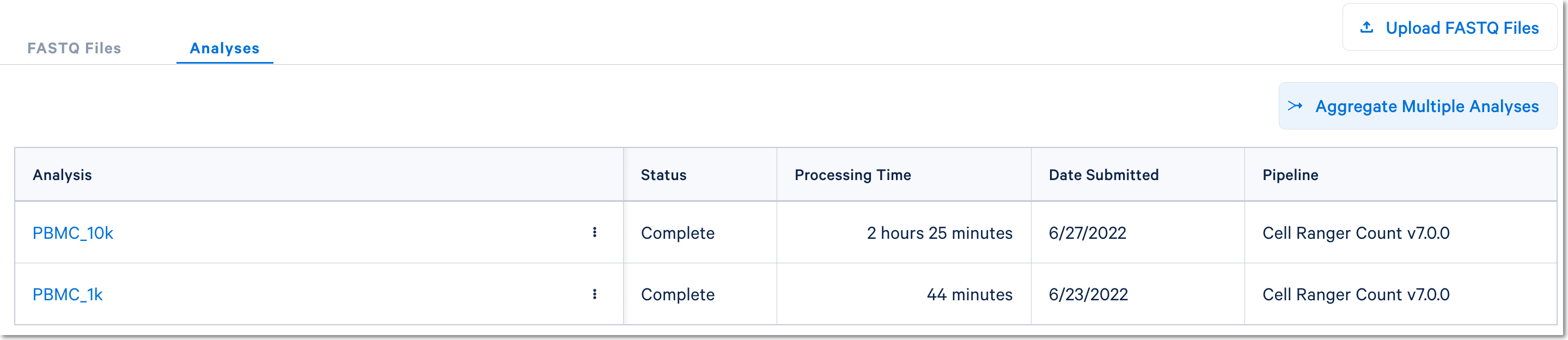
Click the Aggregate Multiple Analyses button to choose the datasets to aggregate and fill out the set up form to run the analysis. The 10x Cloud Analysis set up form autogenerates the aggregation CSV file; users do not need to create one. This file will be available for inspection and download after the analysis has been created.
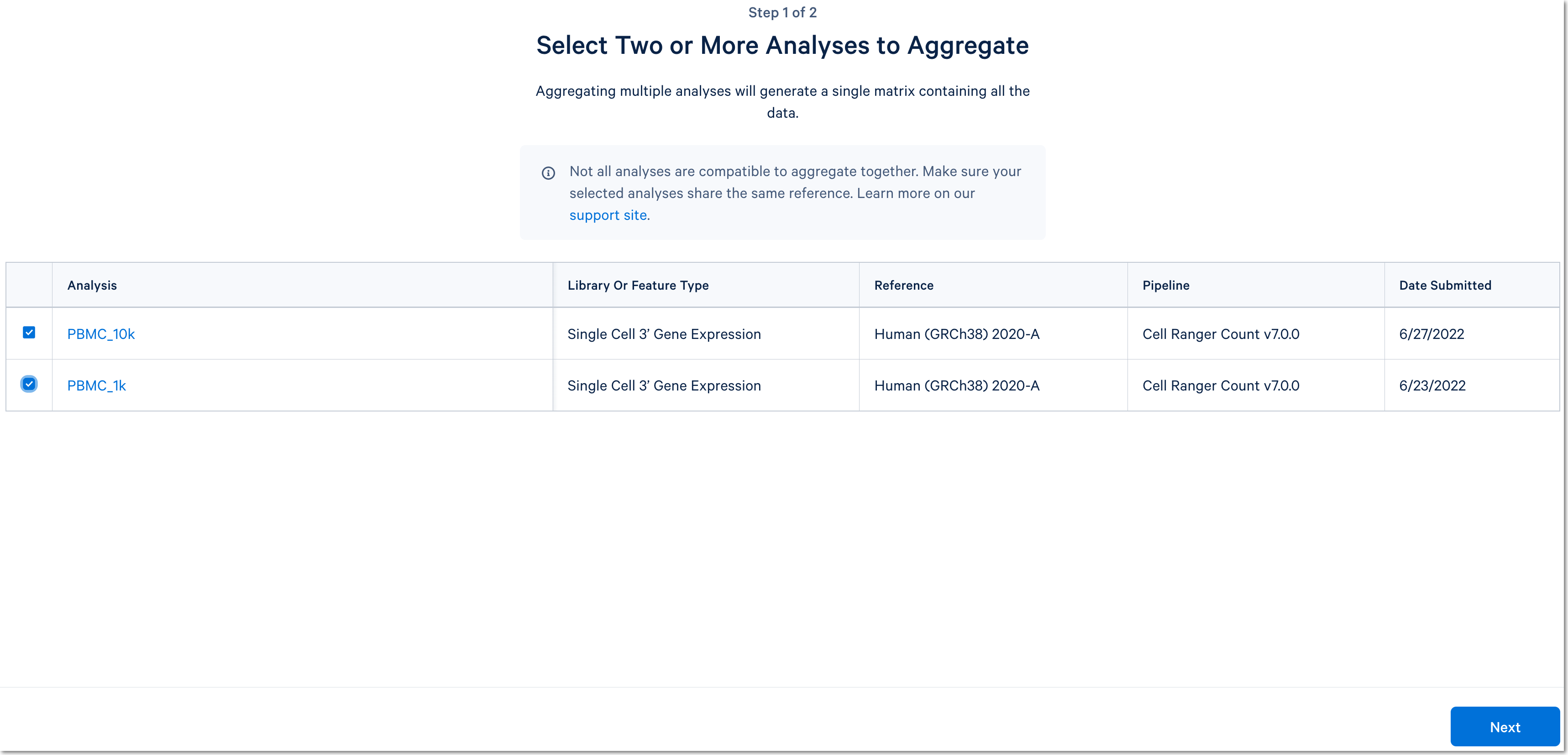
For more detail about this particular example, see this Getting Started tutorial for Cell Ranger aggr (note that the tutorial was written for the command line version of the aggr pipeline).
In this case, there are two libraries - one Gene Expression library and one Multiplexing Capture (CMO) library.
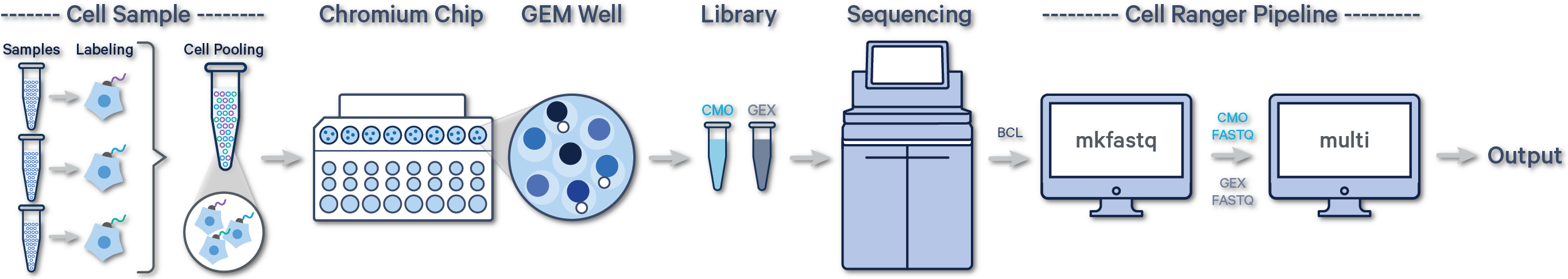
For Cell Multiplexing data, select the FASTQ files corresponding to the Gene Expression library and specify library type as Single Cell 3' Gene Expression. Then, select the FASTQ files for the Multiplexing Capture library and specify feature type as CellPlex Multiplexing Capture. Click Create New Analysis.
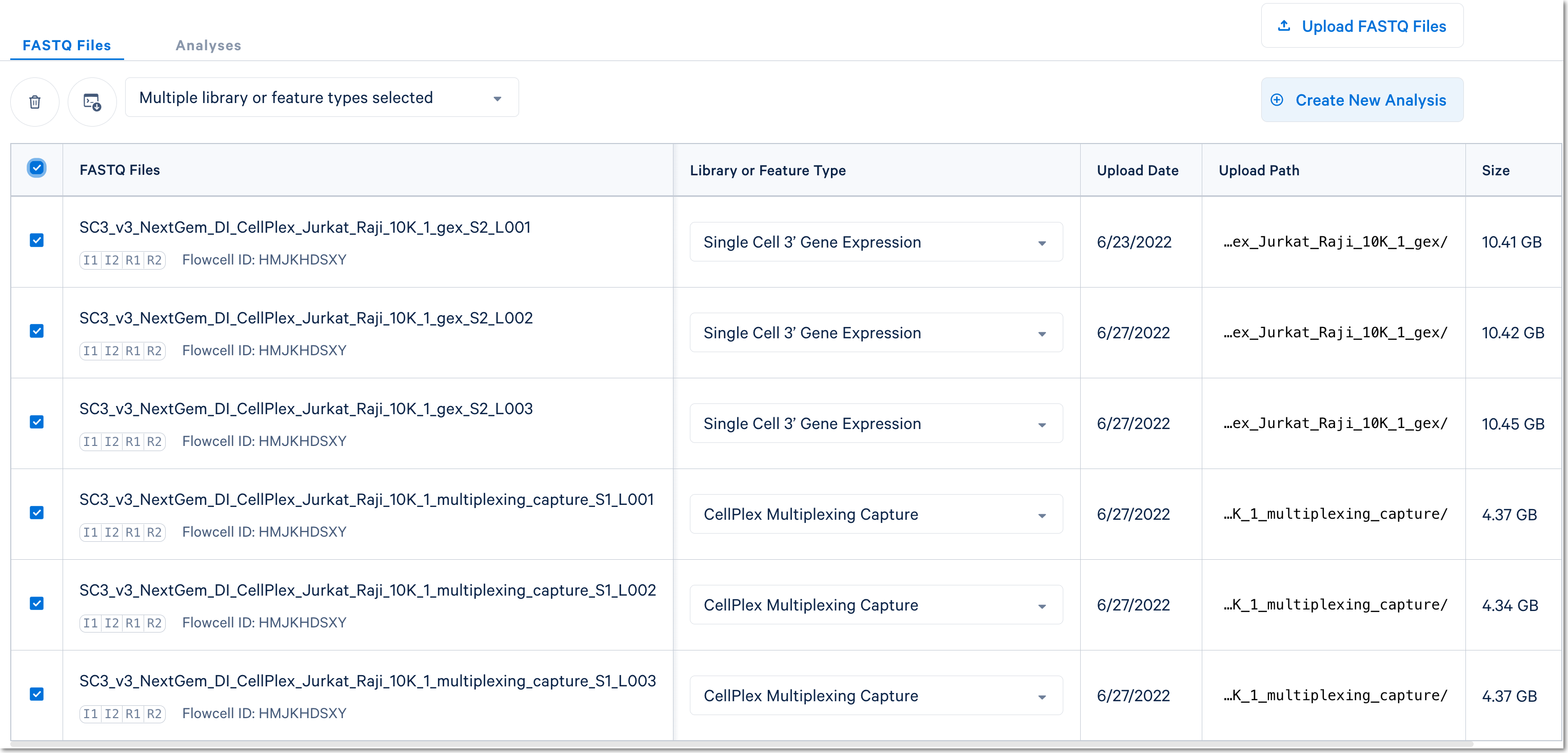
Fill out the setup form to run the analysis. The form autogenerates the multi config CSV file; users do not need to create one. This file will be available for inspection and download after the analysis has been created.
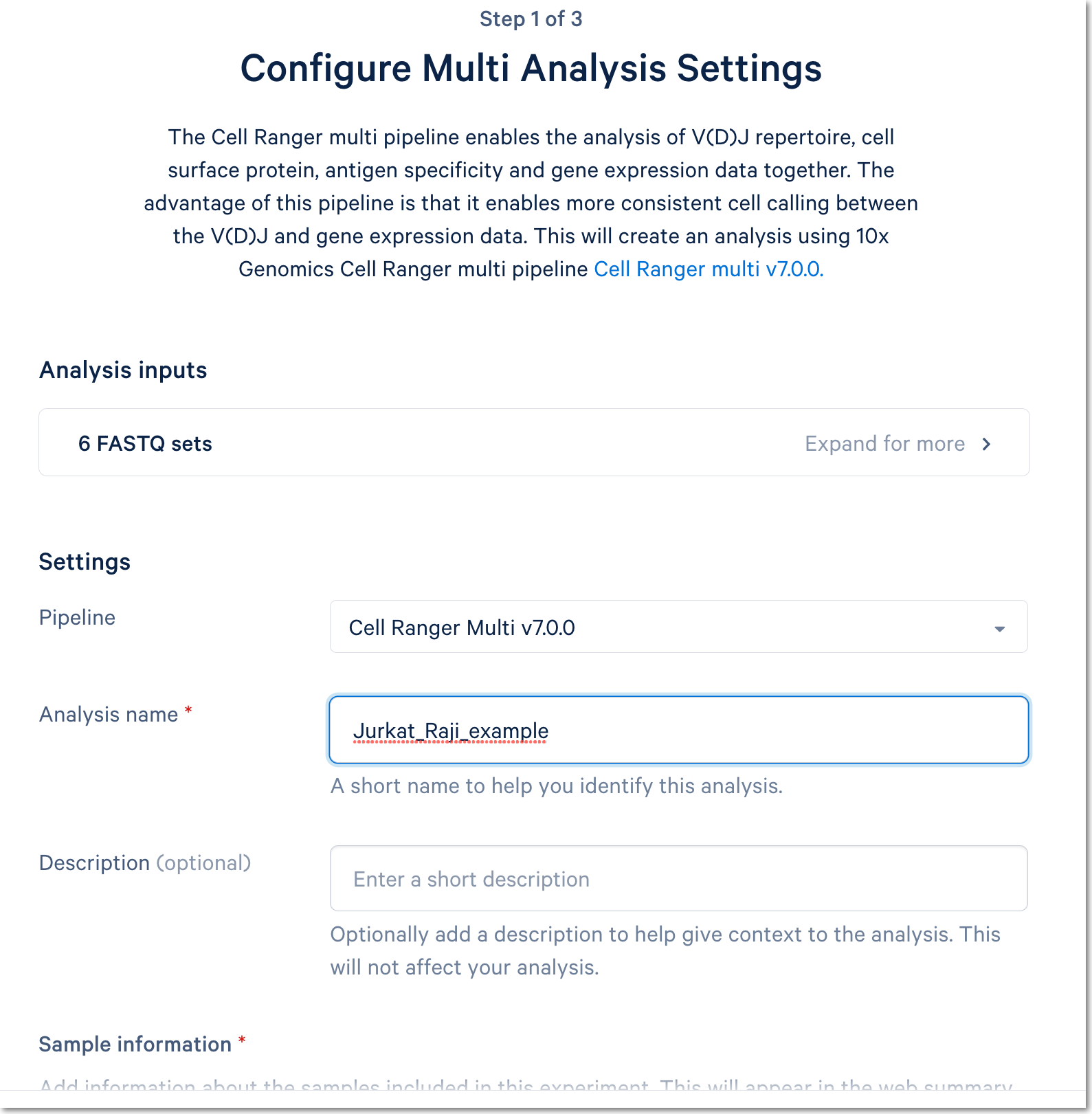
For more detail about this particular example, see this Getting Started tutorial for Cell Ranger multi (note that the tutorial was written for the command line version of the multi pipeline).
As of September 2024, Cloud Analysis introduces a new pipeline for cell type annotation, which can be applied to standard Cell Ranger count and multi outputs to generate cell type labels for human samples. The annotation model is in beta.
Learn how to run the pipeline or learn more about the underlying algorithms and models.
Below is a step-by-step video demonstrating how to create a Cell Ranger analysis on 10x Genomics Cloud Analysis. In this example, one sample was processed in one GEM well and on one flow cell, as illustrated below. After BCL files are converted to FASTQ files (e.g., with Cell Ranger mkfastq or Illumina BCL software), the data can be analyzed with the Cell Ranger count pipeline.
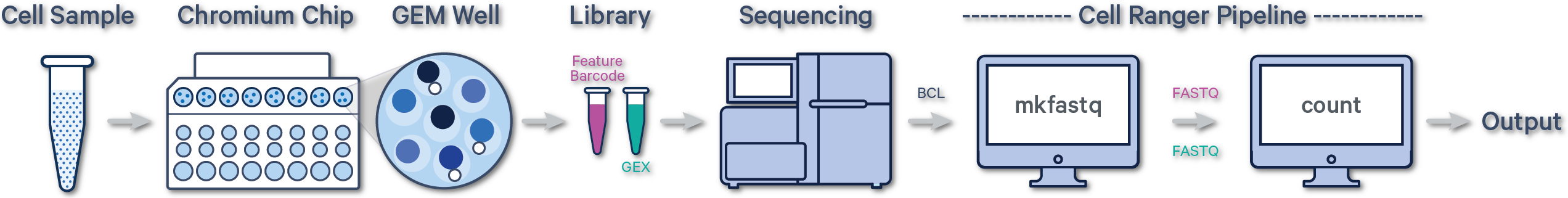
(A single GEM well can yield multiple physical libraries: one Gene Expression library and one or more Feature Barcode libraries. The example below has one Gene Expression library.)